ガラス中に含まれる鉄イオンに起因する 光吸収スペクトルの計算機シミュレーション
Computer Simulation of Optical Absorption Spectra Attributed to Iron Ions in Glass
藤田早苗*・高田章*・土屋博之**・荒井雄介***
Sanae Fujita*, Akira Takada*, Hiroyuki Hijiya**, and Yusuke Arai***
*旭硝子株式会社 先端技術研究所
**旭硝子株式会社 商品開発研究所
***旭硝子株式会社 先進機能ガラス事業本部
ガラス中に不純物として含まれる鉄イオンは、可視~近赤外域の光の吸収源となる。ガラス組成を変更すると鉄イオンの量が同じであっても光吸収スペクトルは変化する。この現象は、鉄イオン周辺の局所構造の変化が引き起こす鉄イオンのd電子の電子状態の変化として理論的に検討することができる。したがって、ガラスの光吸収スペクトルを最適設計するためには、①ガラス組成とガラス中の鉄イオン周辺の局所構造の関係を予測する新技術、および②ガラス中の鉄イオン周辺の局所構造と光吸収スペクトルの関係を予測する新技術の両方が望まれている。
本研究の目的は②の新技術開発を目指したシミュレーション手法の開発である。ガラス中で想定されるような鉄イオン周辺の局所構造の対称性が低い場合にも光吸収スペクトルを効率良く計算できるシミュレーション手法の1つであるDV-ME法を応用することとした。DV-ME法で鉄イオンの光吸収スペクトルの再現性を検討した事例がなかったため、まず様々な局所構造の2価鉄イオンを含む5つの結晶の光吸収スペクトルを計算し、実験結果と比較し、計算精度を確認した。さらに、この手法をガラスに応用展開していく第一段階として、もっとも単純なモデルとして2価鉄イオンを含むシリカガラス構造を作成し、DV-ME法でその構造の光吸収スペクトルを計算し、結晶に関する計算結果と対比しながら分子軌道の観点から局所構造が光吸収スペクトルに与える影響を考察した。
今回検討した2価鉄イオンを含む結晶の光吸収スペクトルに関しては、測定値から-700〜1400 cm-1の誤差はあるものの光吸収ピーク波数の測定値をシミュレーションでほぼ再現でき、また偏光成分の大小関係や光吸収強度に関しても定性的には測定結果とほぼ整合する結果を得ることができた。さらに、鉄イオンのd軌道からなる分子軌道を中継ぎとして、鉄イオン周辺の局所構造と光吸収スペクトルの関係を解明できることもわかった。したがって、本研究により、DV-ME法を用いてガラス中の鉄イオン周辺の局所構造と光吸収スペクトルの関係を予測するためのプロト手法を開発することができた。
Iron ions (Fe2+ and Fe3+) present in glass as impurities cause optical absorption of a range of wavelengths from visible to infrared light. With changes in the glass composition, the molar absorption spectrum associated with the iron ions changes, even if the quantity of iron ion is the same. This causal relation can be examined theoretically on the basis of the changes of electronic state of d-electrons of the iron ion caused by the changes of local structure around iron ion. To optimize absorption spectrum of glass theoretically, two new techniques are required; (i) one is to clarify the relationship among the glass composition and local structure around iron ions, (ii) another is to clarify the relationship among the local structure around iron ions and the resulting absorption spectrum.
In this study, we focus on developing the simulation technique (ii). We use one of the ab-initio simulation methods, DV-ME method. The DV-ME method enables one to efficiently calculate absorption spectrum of glass in which the local structures of iron ions have low symmetry. There has been no previous report on precision of simulated optical absorption spectrum associated with iron ions using the DV-ME method. Hence we first conducted DV-ME calculations of the absorption spectra produced by ferric (Fe2+) ions in seven different local structures, which were cut from five crystals, and then compared measured and simulated wave numbers of the optical absorption peaks produced by Fe2+ ions in these structures. Furthermore, as the first step to apply the DV-ME method to Fe2+ ions in a variety of glasses, we modeled silica-glass structure including Fe2+ ion, and calculated its absorption spectrum, and discussed the influence of local structure of Fe2+ ion on the absorption spectrum, from the viewpoint of molecular orbitals in comparison with those observed in crystals.
The measured wave numbers of the optical absorption peaks produced by Fe2+ ions in crystals were produced with a maximum error of -700〜1400 cm-1. Moreover, measured optical absorption intensity and its polarization-dependence were qualitatively reproduced by this simulation. In addition, we clarified the relationship among the local structure around Fe2+ ions and the resulting absorption spectrum, by analyzing shape of molecular orbitals. In conclusion, we have developed the prototype simulation method for clarifying the relationship among the local structure around Fe2+ ions and the resulting absorption spectrum either in crystals and glasses.
1. 緒言
1.1. Feイオンによる光吸収
ソーダライムガラスは、高い可視光透過率、耐候性、耐熱性を有し、また量産が容易なため、自動車や住宅の開口部材や光学部材に広く用いられてきた。ソーダライムガラスの主成分はSiO2, (Mg, Ca)O, (Na, K)2Oであるが、ガラスの製造工程において、10万原子中に数個程度のFeイオンが製造装置や原料由来の不純物として混入する。Feイオンはガラス中でFe3+あるいはFe2+として存在し、それぞれ可視域あるいは可視~近赤外域の、特定の波長の光の吸収源になることが知られている(1)。従来では不純物レベルのFeイオンによる光吸収が実用上の問題となることは殆どなかった。しかし近年の太陽熱発電用途の量産ガラスでは、不純物レベルのFeイオンの可視~近赤外域での光吸収の低減が一層求められるようになっている。
Feイオンによる光吸収スペクトルはガラス組成により変化することが実験的にわかっている。Feイオン低減に限界があるため、実用上はガラス組成を調整することで発電効率が向上するような光吸収スペクトルに近づけている(2)(3)。配位子場理論(4)を用いると、ガラス組成による光吸収スペクトルの変化は、Feイオン周辺の局所構造の変化に伴う、Feイオンのd電子の電子状態の変化として検討することができる。ここでいう局所構造とは、Feイオンの電子状態に大きな影響を与える半径2~5 Å程度の範囲の原子の立体構造を想定している。もしガラス組成、ガラス中のFeイオン周辺の局所構造および光吸収スペクトルの間の関係をさらに定量的に解明できれば、ガラス組成の調整による光吸収スペクトルの制御を効率的に行うことができるようになる。
ガラス中のFeイオンは、X線吸収分光(XAS)等の測定により、4, 5, 6個の酸素イオンに取り囲まれており(すなわち4, 5, 6配位になる)、その周辺は対称性の低い局所構造になっていると考えられている。ソーダライムガラス中のFeイオン周辺の局所構造の組成による定性的な変化は、XAS(5)(6)(7)や共鳴非弾性X線散乱(RIXS)、ESR(8)といった測定により推測されている。しかし、Fe-O距離およびO-Fe-O角度等の、詳細な局所構造や配位数分布を測定だけで決定することは困難である。
Feイオン周辺の局所構造の対称性が高い場合には、配位子場理論により光吸収波数を解析的に求めることができ、経験的パラメータの調整により測定値を再現することができる。たとえばガラス中のFe2+イオンの場合、配位子場理論の結果を援用し、5000 cm-1付近を4配位、9000 cm-1付近を6配位とする帰属が知られている(9)(Fig. 1)。しかしながら、ガラス中で想定されるような対称性の低い局所構造の場合に、高い対称性を仮定して経験的に近似する手法では、部分的にしか光吸収スペクトルを説明できないと推定される。
したがって、ガラス中のFeイオン周辺の局所構造と光吸収スペクトルの関係を定量的に議論することは現在でも難しい課題である。
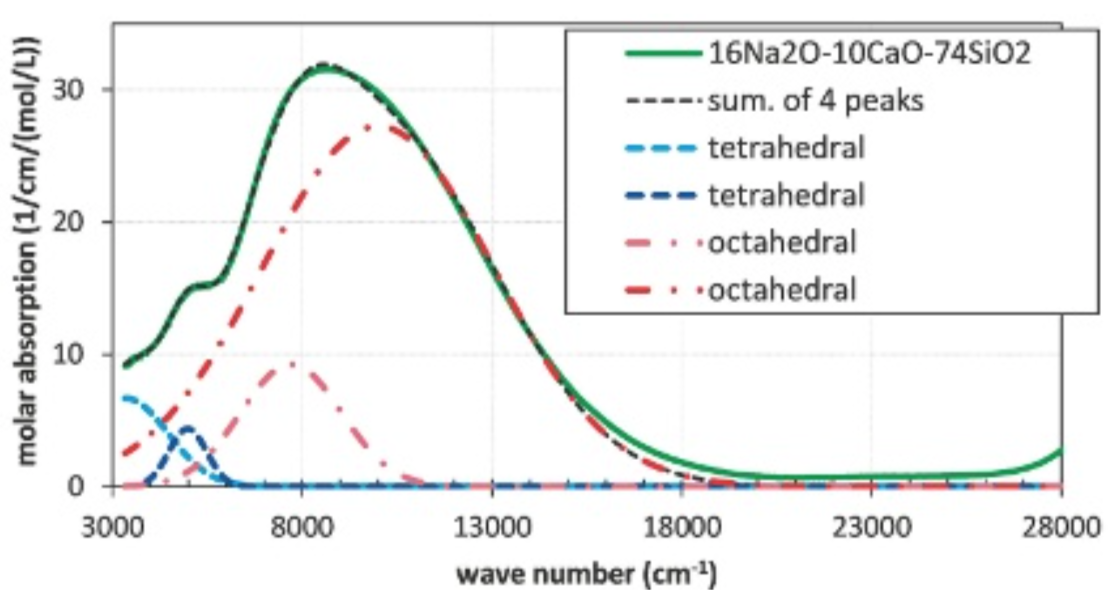 Fig.1 Absorption spectrum of Fe2+ ion included in soda- lime glass doped with 0.16 mol % Fe2O3 and assignment of the peaks following as Volotinen(9). Two peaks are assigned for Jahn–Teller distorted octahedral and tetrahedral sites respectively.
Fig.1 Absorption spectrum of Fe2+ ion included in soda- lime glass doped with 0.16 mol % Fe2O3 and assignment of the peaks following as Volotinen(9). Two peaks are assigned for Jahn–Teller distorted octahedral and tetrahedral sites respectively.1.2. 本研究の目的
測定だけでは詳細を決定することが困難なFeイオン周辺の局所構造と、Feイオンの光吸収スペクトルの関係を、局所構造の対称性を問わず経験的パラメータを用いずに原子や電子レベルのシミュレーションで精度よく解析できれば、ガラス中のFeイオン周辺の局所構造と光吸収スペクトルに関する重要な知見が得られる。経験的パラメータを用いずにFeイオンの光吸収計算を様々な構造について行った例は筆者の知る限り報告されていない。そのため我々は、構造が既知である結晶中や、SiO2ガラス構造モデル中のFeイオンについて光吸収スペクトルのシミュレーションを実行し、計算精度を確認しながら、局所構造と光吸収スペクトルの関係解明を試みることとした。
Fe2+イオンは、Fe3+イオンよりも可視~近赤外域でのモル吸光度が大きいことから、太陽熱発電用途のガラスではFe2+イオンによる光吸収スペクトルの調整が重要視されている。そのため本稿ではFe2+イオンの解析に注力した。なおFe3+イオンによる可視光吸収強度が弱い理由は、遷移の前後でスピン多重度が変化するため(「スピン禁制遷移」と呼ばれる現象)である。スピン禁制遷移の励起状態に、スピン軌道相互作用を通して基底状態と同じスピンを持つ状態が混合する場合、部分的に許容遷移となり光吸収強度を得る。シミュレーション時にスピン軌道相互作用を考慮することで、Fe3+イオンについても今回のFe2+によるスピン許容遷移の光吸収スペクトル評価とほぼ同様の手順が適用できる。
本稿では、2.1節で配位子場理論について概要を説明した後に、2.2節で配位子場理論に基づく多重項計算を紹介し、2.3節で第一原理計算の一手法であり本研究で採用したDV-ME法による光吸収スペクトルの計算手順の概要を説明する。3.1節および3.2節では不純物の影響がみられないと考えられるケイ酸塩もしくは炭酸塩からなる6結晶(ferrosilite, fayalite, siderite, gillespite, almandine, staurolite)中の計8種類のFe2+イオンの局所構造モデルにおける、光吸収スペクトルのDV-ME法計算結果と測定結果の対比を報告する。これらの結晶中ではFe2+イオンの酸素配位数(4, 6, 8)や配位多面体の歪みは様々である。したがってガラス中で様々な配位数(4, 5, 6)や対称性の局所構造をとると考えられているFe2+イオンの光吸収スペクトルも同程度の精度で評価できると期待できる。3.3節でSiO2ガラス構造モデル中のFe2+イオンの光吸収スペクトルのDV-ME法計算結果を述べる。不純物の影響のため実績対比できていないが、ガラス中で存在が示唆されている5配位のFe2+イオンを含む数少ない結晶であるgrandidieriteや、歪んだ4配位のFe2+イオンを含むpellyiteの計算結果も3.3節で紹介する。4章で考察を述べる。
2. Feイオンの光吸収スペクトルの計算手法
2.1. 配位子場理論
金属イオンを取り囲む陰イオンを配位子(ligand)と呼び、配位子が金属イオンに与える影響を配位子場、配位子の数を配位数、配位子を頂点とする多面体を配位多面体という。
20世紀前半に誕生した結晶場理論によると、自由イオン中で5重縮退するd軌道は、配位子に囲まれると配位子場の対称性に応じ分裂する。例えばOh対称(Fig. 2(a))の配位子場におかれた場合には、各d軌道の形状に応じてエネルギー差⊿oの2状態t2gおよびegに分裂する(Fig. 3)。中心の遷移金属イオンと配位子との距離が短くなると配位場が強くなり⊿oは大きくなる。⊿oが十分小さいとき基底状態はスピンの向きがなるべく揃った状態となり、Fe2+の基底状態では5個のd電子は上向きスピン、1個のd電子は下向きスピンを持つ。t2gにある1つの下向きスピンがegにスピン許容遷移することで、波数⊿oの光が吸収される。この遷移の前後での電子状態は、スピン多重度と空間部分の対称性を表す記号を用いて、5T2gおよび5Egなどと書かれる。Td対称(Fig. 2(b))の場合も同様に、エネルギー差⊿tの2状態間でスピン許容遷移が生じる(Fig. 3)。配位子場の対称性がOhおよびTdから低下するに従い、d軌道は3~5つに分裂する。したがって結晶およびガラス中で1つのFe2+イオンからスピン許容遷移による光吸収ピークは最大で4本生じる。
20世紀半ばに誕生した配位子場(Ligand Field)理論(以下LF理論)を用いると、配位子場を点電荷として扱う結晶場理論と異なり、配位子と遷移金属イオン間の軌道の混成を考慮したd軌道の分裂が検討できる。特に、配位子場がOh対称の場合、⊿oなどの経験的パラメータを用いることで、2~8個のd電子を持つ遷移金属イオンについて光吸収波数が解析的に得られており、「田辺-菅野ダイヤグラム」として知られている。Td対称の4配位の配位子場におかれたn個のd電子を持つ遷移金属イオンの場合には、空孔の遷移と考えることで、(10-n)個のd電子の田辺-菅野ダイヤグラムが適用できる(4)。
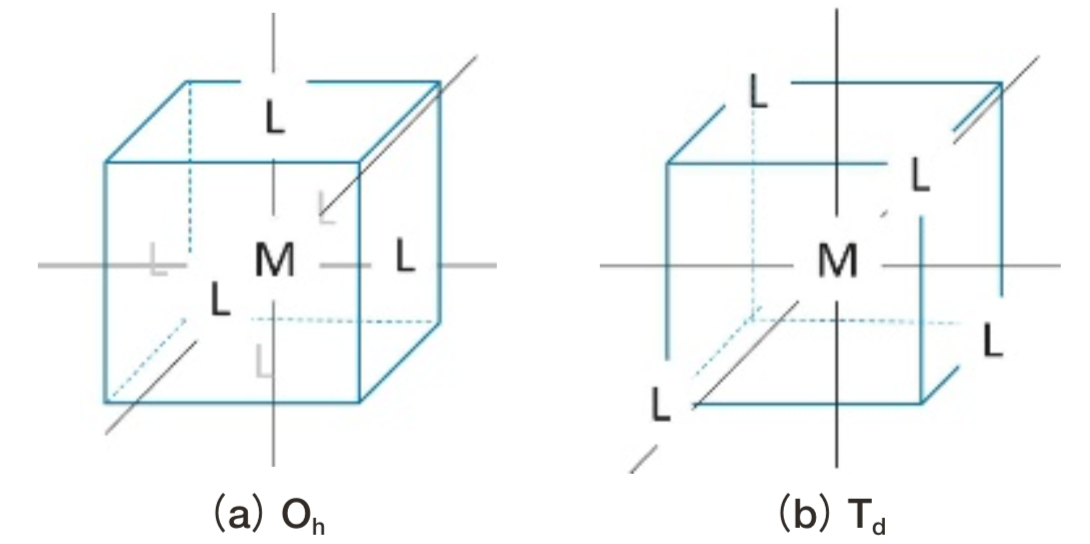 Fig.2 Coordination polyhedra with cubic symmetry. M represents transition metal ion. L represents ligand. Ligands are located at face-centered cites and at the alternative vertices for Oh and Td symmetry, respectively.
Fig.2 Coordination polyhedra with cubic symmetry. M represents transition metal ion. L represents ligand. Ligands are located at face-centered cites and at the alternative vertices for Oh and Td symmetry, respectively.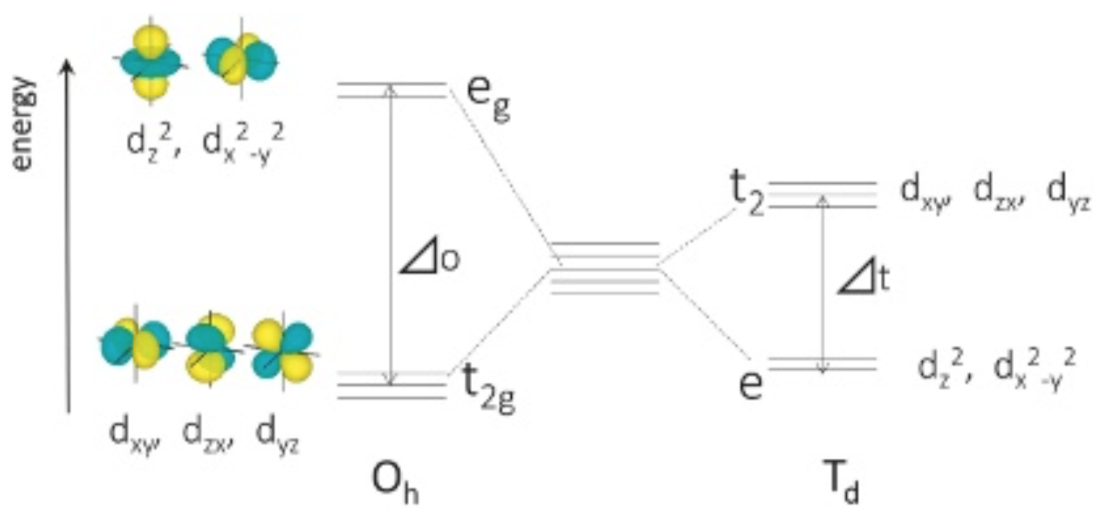 Fig.3 Energy levels of d-orbitals in octahedral and tetrahedral ligand field. ⊿ represents strength of ligand field. ⊿ value not only changes depending upon ligand species , but also increases as ligand- metal distance becomes shorter.
Fig.3 Energy levels of d-orbitals in octahedral and tetrahedral ligand field. ⊿ represents strength of ligand field. ⊿ value not only changes depending upon ligand species , but also increases as ligand- metal distance becomes shorter.1章で述べたように、ガラス中でFeイオン周辺の局所構造は歪んでおりOhあるいはTd対称と見なし難い構造になっていると考えられている。そのためガラス中のFeイオンによる光吸収スペクトルを田辺-菅野ダイヤグラムだけを用いて定量的に説明することには限界がある。
2.2. LF理論に基づく多重項計算
結晶中でOhあるいはTd対称(Fig. 2)の配位子場を受けるFeイオンついて、XASのK端のpre-edgeスペクトルのLF理論に基づく多重項計算が2000年に報告されている(10)。その後、扱える対称性はC3v(Fig. 4(a))といった5配位を含むものに拡張され(11)、2016年にはVercamerらにより結晶中Feイオンの光吸収スペクトルおよびXASがほとんど等価な経験的パラメータセットで記述できることが報告されている(12)。ただし対称性が低くなるほど配位子場の強さを表すために必要な経験的パラメータ数が増え評価が煩雑になるため、Vercamerら(12)はFig. 4に一例を示すように高い対称性へ近似している。
LF理論に基づく多重項計算は、ある程度高い対称性を経験的に仮定する必要があるが、詳細な構造を決定する必要がなく簡便に計算ができるという利点がある。経験的パラメータの分布を仮定することで、ソーダライムガラスのFe2+イオンによる光吸収スペクトルの測定結果を、5配位による光吸収スペクトルの重ね合わせとして再現できることがVercamerらにより示されている(8)。従来の多くの文献ではガラス中Fe2+イオンの光吸収スペクトルを4配位と6配位のみの重ね合わせで説明しており、5配位のみで測定および理論に整合する説明をした初めての例である。
今後、ガラス組成による経験的パラメータの変化を評価することができれば、ガラス組成とFeイオンの局所構造と光吸収スペクトルの間の関係を把握できるものと期待される。
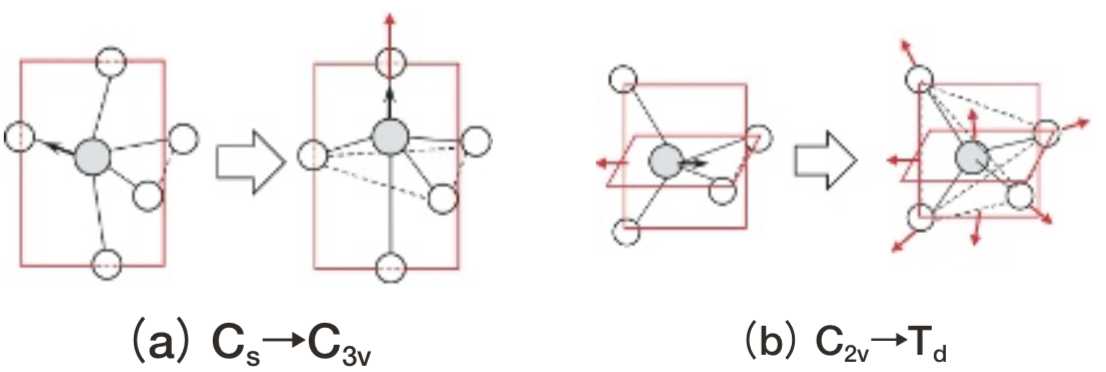 Fig.4 Schematic diagram of structural changes from lower symmetry to higher symmetry in grandidierite(a), and staurolite(b). Part of symmetry operations are indicated by red planes for mirror and arrows for rotation.
Fig.4 Schematic diagram of structural changes from lower symmetry to higher symmetry in grandidierite(a), and staurolite(b). Part of symmetry operations are indicated by red planes for mirror and arrows for rotation.2.3. DV-ME法計算
20世紀後半、原理的には構造の対称性を問わず経験的パラメータが不要な第一原理計算が誕生し、電子遷移による光吸収を扱うのに適した理論も開発されている。第一原理計算では、基底や擬ポテンシャル、交換相関相互作用といった部分では近似理論が用いられており、それぞれに複数の選択肢がある。また、励起状態の計算手法には、配置換相互作用法や時間依存密度汎関数法など、複数の手法が存在する。第一原理計算を用いてFeイオンの光吸収と局所構造の関係を定量的に議論した先行研究はなく、どのような近似理論や計算手法が適しているかは知られていない。
第一原理計算による遷移金属イオンの光吸収スペクトル計算が報告されている例は、筆者の知る限りは、関西学院大学の小笠原らにより開発されたDV-ME(discrete-variational multi-electron)法(13)によるものである。ルビー(Cr3+:α-Al2O3)で良好な再現性が報告されている(14)ことから、我々はDV-ME法を採用した。
DV-ME法は多電子系の波動関数やエネルギーを解析する計算手法である。一電子近似での電子状態を解析するDV-Xα法(15)をベースとして開発され、DV-Xα法と同様に数原子~数十原子からなるクラスターモデル(Fig. 5)を用いる。クラスターモデルを取り囲むすべてのイオンによる影響を点電荷でモデル化することで、固体や結晶を近似できる。
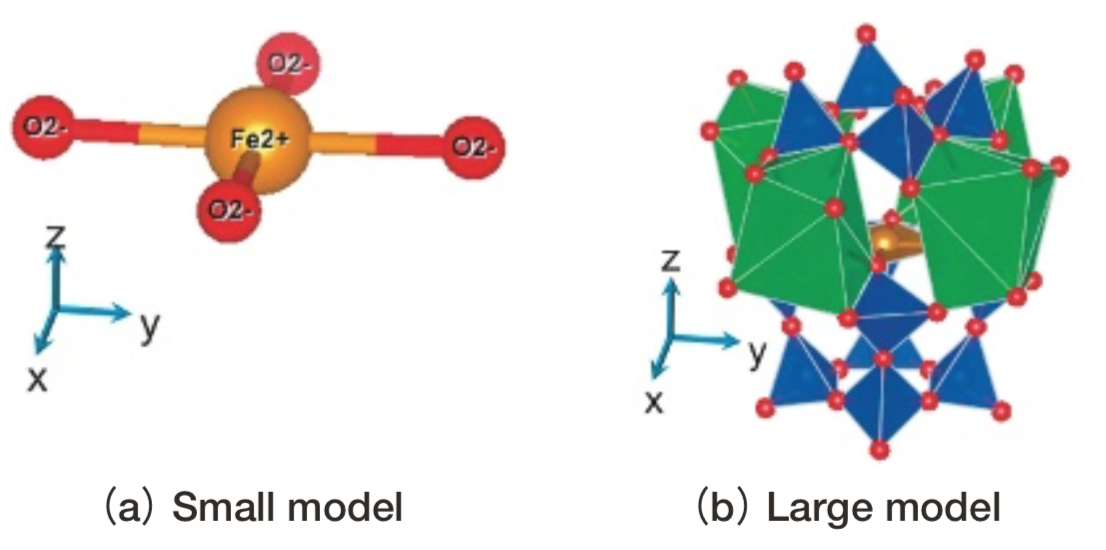 Fig.5 Two cluster models for gillespite. Small model consists of only one coordination polyhedron. Large one consists of several polyhedra. Point charges around cluster are not drawn for the ease of viewing. Red ball: O2-. Brown ball: Fe2+. Blue polyhedron: SiO4. Green polyhedron: BaO8.
Fig.5 Two cluster models for gillespite. Small model consists of only one coordination polyhedron. Large one consists of several polyhedra. Point charges around cluster are not drawn for the ease of viewing. Red ball: O2-. Brown ball: Fe2+. Blue polyhedron: SiO4. Green polyhedron: BaO8.DV-ME法計算の手順をFig. 6および以下に示す。第一ステップとして、初期電荷に形式電荷を与え一電子計算を行う。今回は一電子計算としてDV-Xα法を採用し、交換ポテンシャルにはα=0.7としたXαポテンシャルを用いた。次に第二ステップとして、一電子計算結果をもとにFe2+イオンのd軌道を主成分に含むような5個の分子軌道の配置間相互作用を考慮して多重項計算を行う。得られた多重項に対して、単位ベクトルに平行な電場振動面を持つ光の光吸収強度Iを式(1)により計算し、適当な関数で拡幅することでスペクトルを求める。以下の計算結果はすべて半値半幅0.02eVのガウス関数を利用する。
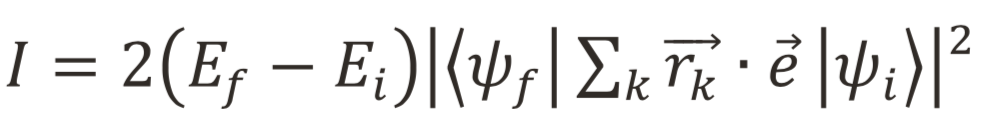 (1)
(1)ただし、,はそれぞれ基底状態および光吸収後の終状態の多電子波動関数、EiとEfはそれぞれ基底状態および終状態のエネルギー、はk番目の電子の位置ベクトルを示す。
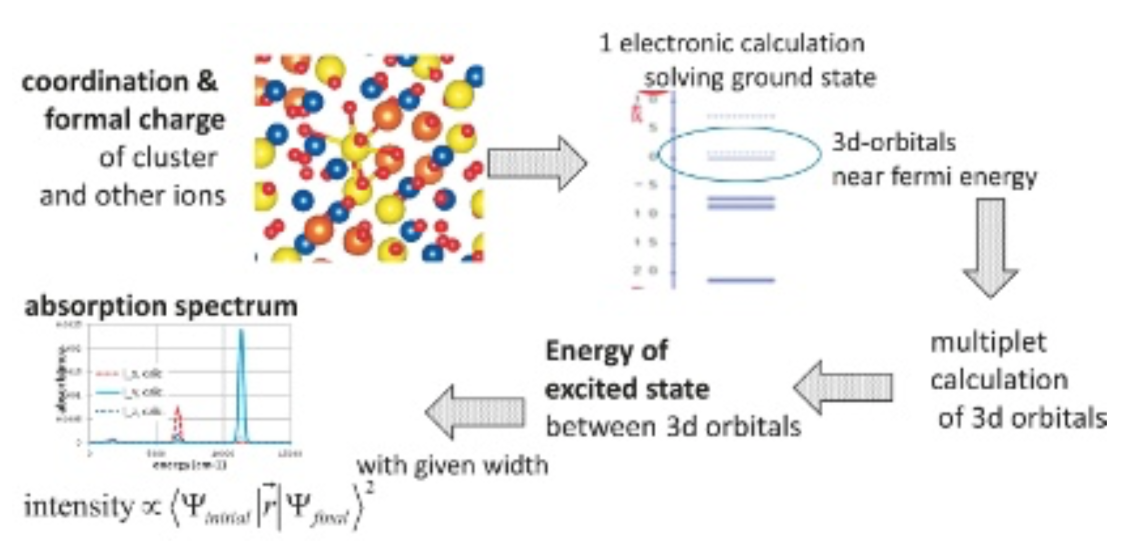 Fig.6 Schematic flow chart of DV-ME method.
Fig.6 Schematic flow chart of DV-ME method.3. DV-ME法計算によるシミュレーション結果
3.1. 結晶中Fe2+イオンの光吸収スペクトルの再現性
Fe2+イオンは結晶中のMgやAl, Ca等の他種イオンのサイトに僅かに置換することがある。置換後の局所構造の実測データは得られなかったため、今回は置換のない結晶中Fe2+イオンについてこれらの光吸収スペクトルをDV-ME法計算で評価し、どの程度定量的に再現できるか確認した。Table 1に今回取り上げた結晶中Fe2+イオン周辺の局所構造の対称性や配位数、歪の大きさの指標を示す。
計算に用いたモデルは、小規模モデル(Fig. 5(a))でクラスターを取り囲むイオンを点電荷として考慮したものとしないもの、あるいは大規模モデル(Fig. 5(b))でクラスターを取り囲むイオンを点電荷として考慮したものの計3種である。クラスターを取り囲む点電荷を考慮したもの、あるいは大規模モデルの方が、結晶中のFe2+イオン周辺の局所構造をより正確に表現していることになる。以下の計算では特に断らない限り、クラスターを取り囲む点電荷を考慮するものとする。その他の計算条件の詳細は文献 (16)に報告している。
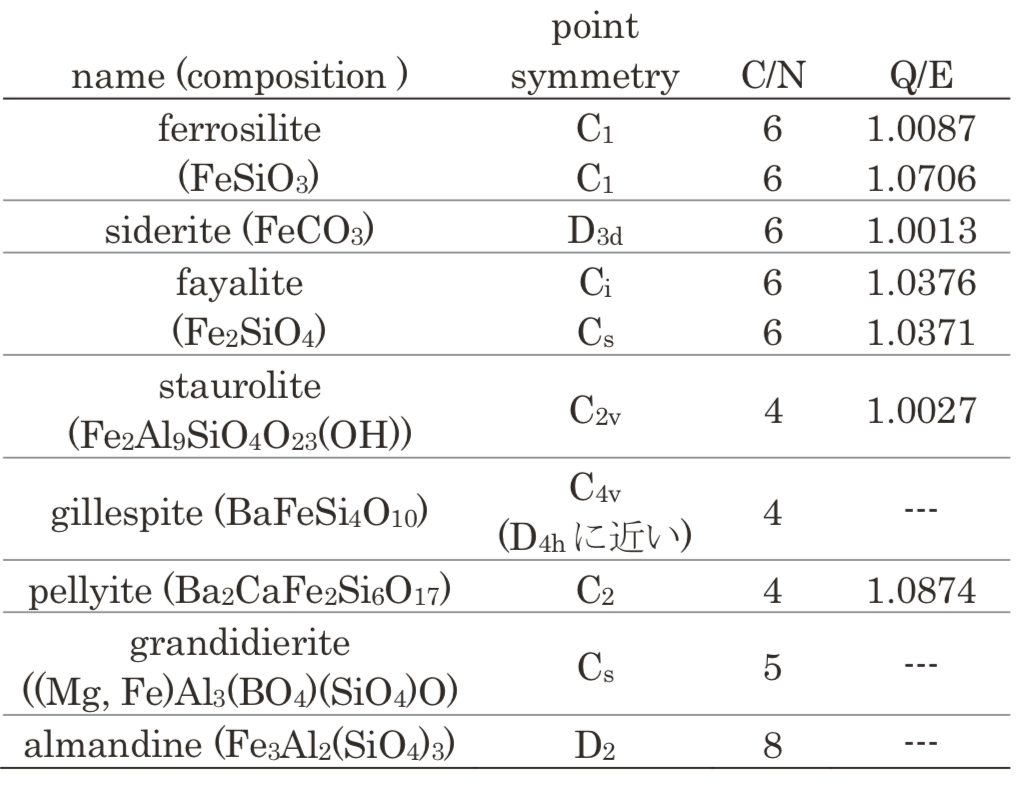 Table 1 Characters of Fe2+ polyhedra in simulated crystals.
C/N: coordination number, Q/E: quadratic elongation1
Table 1 Characters of Fe2+ polyhedra in simulated crystals.
C/N: coordination number, Q/E: quadratic elongation1本稿では、すべての原子位置が測定で決定され、かつ不純物による影響がみられないと考えられる光吸収スペクトルの測定結果が得られているferrosilite, fayalite, sideriteおよびgillespiteの光吸収スペクトル計算結果と測定結果の対比を詳しく示す。Ferrosilite, fayalite, sideriteは後述する理由のため大規模モデルでは計算しておらず、小規模モデルによる計算のみ示す。gillespiteについては大規模モデルの計算結果も示す。これら4種以外の結晶staurolite, pellyite, grandidierite, almandineについては、詳細を掲載しなかった理由および計算結果の概要を3.1.5項で紹介する。
以下では、光吸収強度の計算結果を、spinel中の4配位Mgイオンに置換したFe2+イオンの小規模モデルの光吸収強度が1となるように規格化する。0.1未満のものは非常に小さい吸収とみなせる。
3.1.1. Ferrosilite
Ferrosiliteの単位胞と2種のFe2+イオンのサイトM1, M2に対応する小規模モデルをFig. 7に示す。M1, M2サイトの配位多面体は結晶中では辺共有しながらb-c平面内で無限に連なっている。
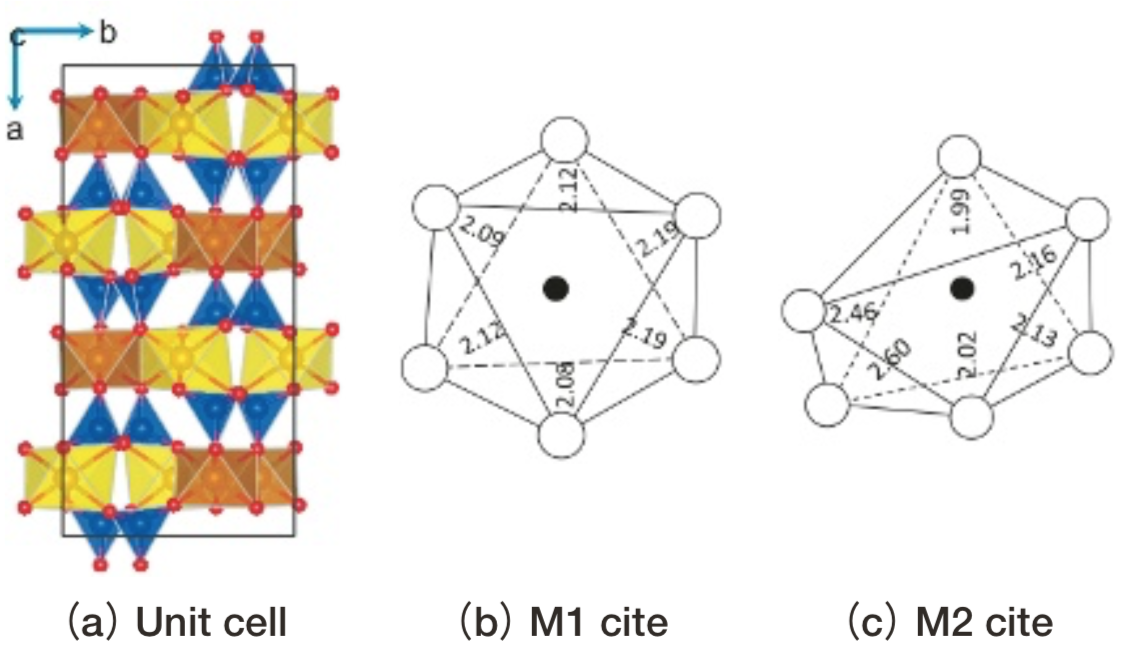 Fig.7 Structure of ferrosilite(FeO-SiO2). Blue polyhedron: SiO4, Brown or yellow polyhedron: FeO6 .
Fig.7 Structure of ferrosilite(FeO-SiO2). Blue polyhedron: SiO4, Brown or yellow polyhedron: FeO6 .Fig. 8にferrosiliteのM1, M2サイトの光吸収スペクトルの計算結果を測定データ(17)(18)とともに示す。主要な測定ピーク(Fig. 8(a)中a, b, d)に関して、1000 cm-1程度の精度で光吸収波数を、また各ピークの偏光の最大成分を、小規模モデルを用いて再現できている。なお大規模モデルで第一ステップである一電子計算をすると、配位多面体を辺共有する周辺Fe2+イオンのd軌道との混成が大きく結晶中ではバンドを形成すると考えられることから、クラスターモデルでは正確な配置間相互作用の計算が困難と判断した。そのためferrosiliteの大規模モデルでは第二ステップであるDV-ME法計算は行っていない。
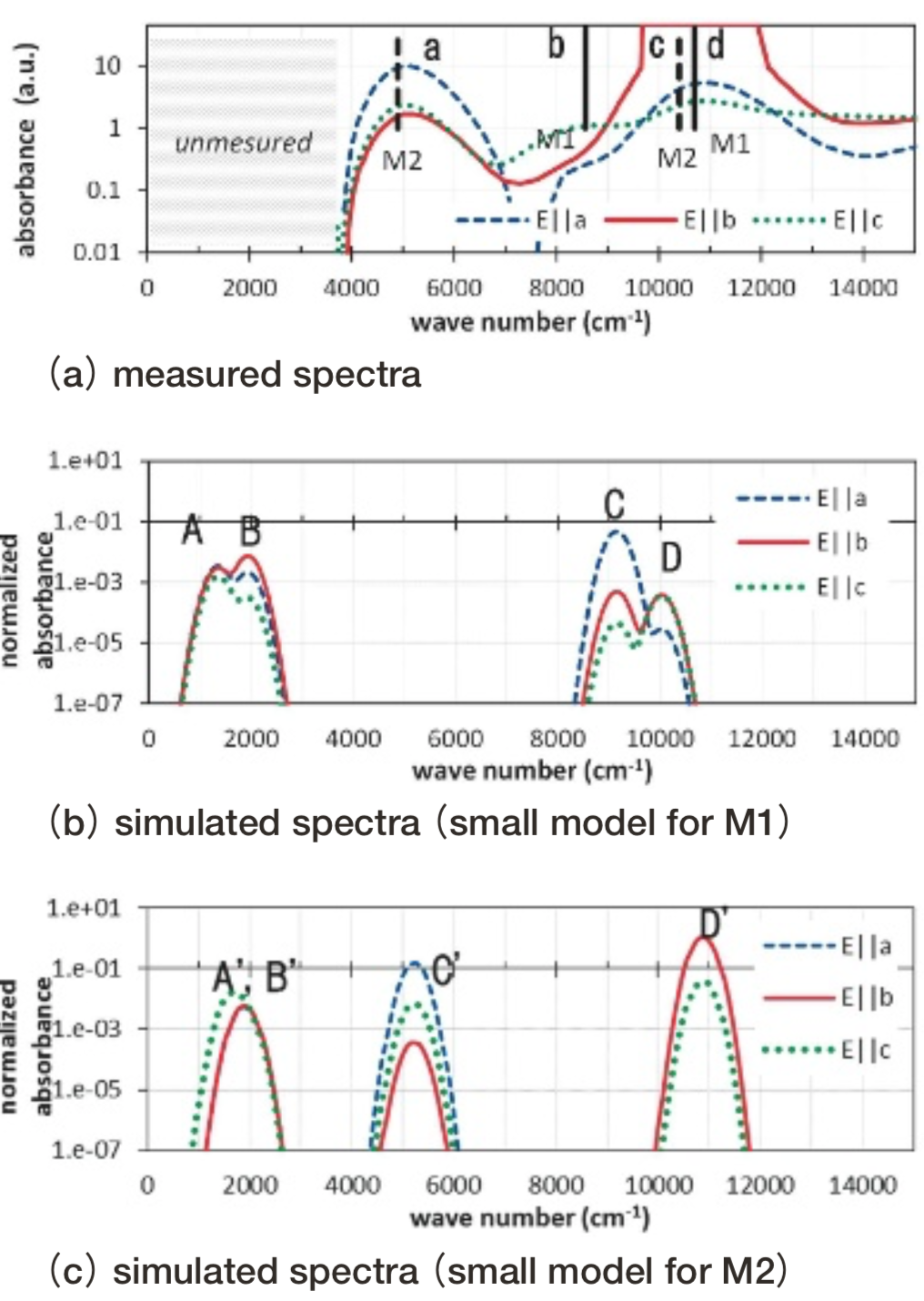 Fig.8 Optical absorption spectra of ferrosilite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns tobe 1. Line sin (a): measure d spectra of hypersthene(Mg0.60, Fe0.40)O-SiO2)(17)which is a solid solution of enstatite and ferrosilite. Vertical solid and dashed bars in(a): peak maxima in ferrosilite assigned to M1 and M2 cite respectively.
Fig.8 Optical absorption spectra of ferrosilite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns tobe 1. Line sin (a): measure d spectra of hypersthene(Mg0.60, Fe0.40)O-SiO2)(17)which is a solid solution of enstatite and ferrosilite. Vertical solid and dashed bars in(a): peak maxima in ferrosilite assigned to M1 and M2 cite respectively.3.1.2. Fayalite
Fayaliteの単位胞とM1, M2サイトの小規模モデルをFig. 9に示す。M1, M2サイトの配位多面体は結晶中では辺および点共有しながら3次元的に無限に連なっており、先述したferrosiliteの場合と同様の理由で、大規模モデルの計算を行っていない。
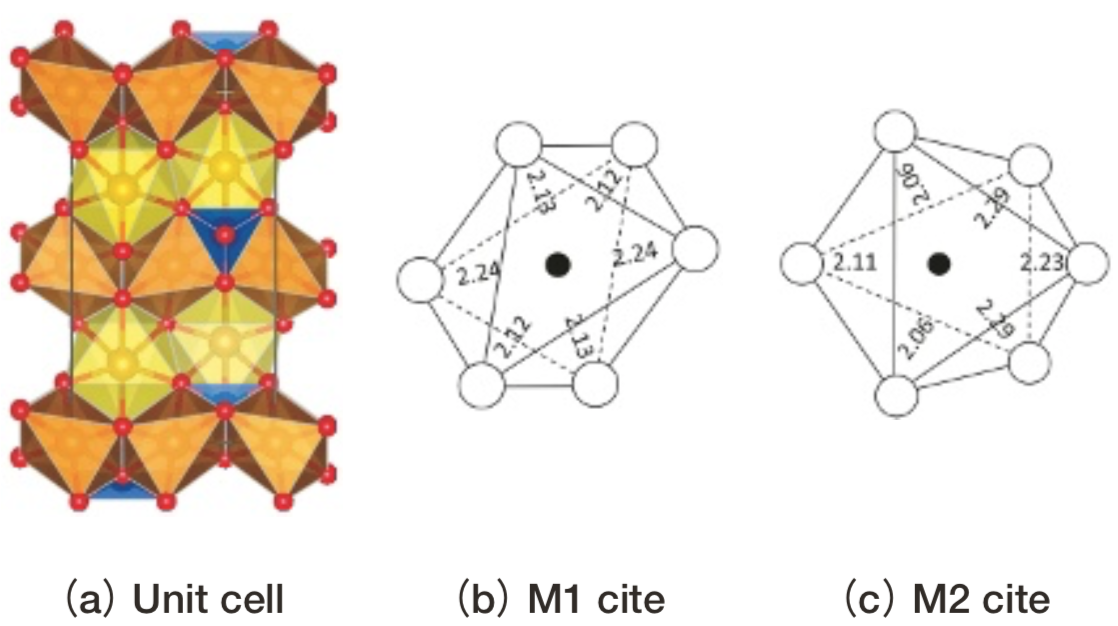 Fig.9 Structure of fayalite projected onto(100).(a)shows the unit cell. Blue polyhedron: SiO4, Brown or yellow polyhedron: FeO6 . (b)and(c)show small models for M1 cite and M2 cite respectively. ●: Fe2+, ○:O2-. Fe-O distances in each site are indicated(Å).
Fig.9 Structure of fayalite projected onto(100).(a)shows the unit cell. Blue polyhedron: SiO4, Brown or yellow polyhedron: FeO6 . (b)and(c)show small models for M1 cite and M2 cite respectively. ●: Fe2+, ○:O2-. Fe-O distances in each site are indicated(Å).Fig. 10にfayaliteのM1, M2サイトの光吸収スペクトルの計算結果を測定データ(17)(18)とともに示す。M1サイトは反転対称を持ち規格化された光吸収強度の計算値は1e-05未満と非常に小さく誤差の範囲で0とみなせるが、測定では振電相互作用により光吸収強度を得ているものと考えられている。ピークA, B, A’, B’は測定波数の下限値以下にあり、測定との対比はできていない。M1サイトに測定で帰属されている2本のピークにはピークC, Dが対応すると考えられる。M2サイトには測定では1本のピークが帰属されているが、計算では測定波数域にC’, D’の2本のピークが得られている。偏光依存性からピークD’が測定のピークに対応すると考えると、光吸収波数は1000 cm-1未満で整合する。ピークC’は測定に対応するものがあるか不明である。
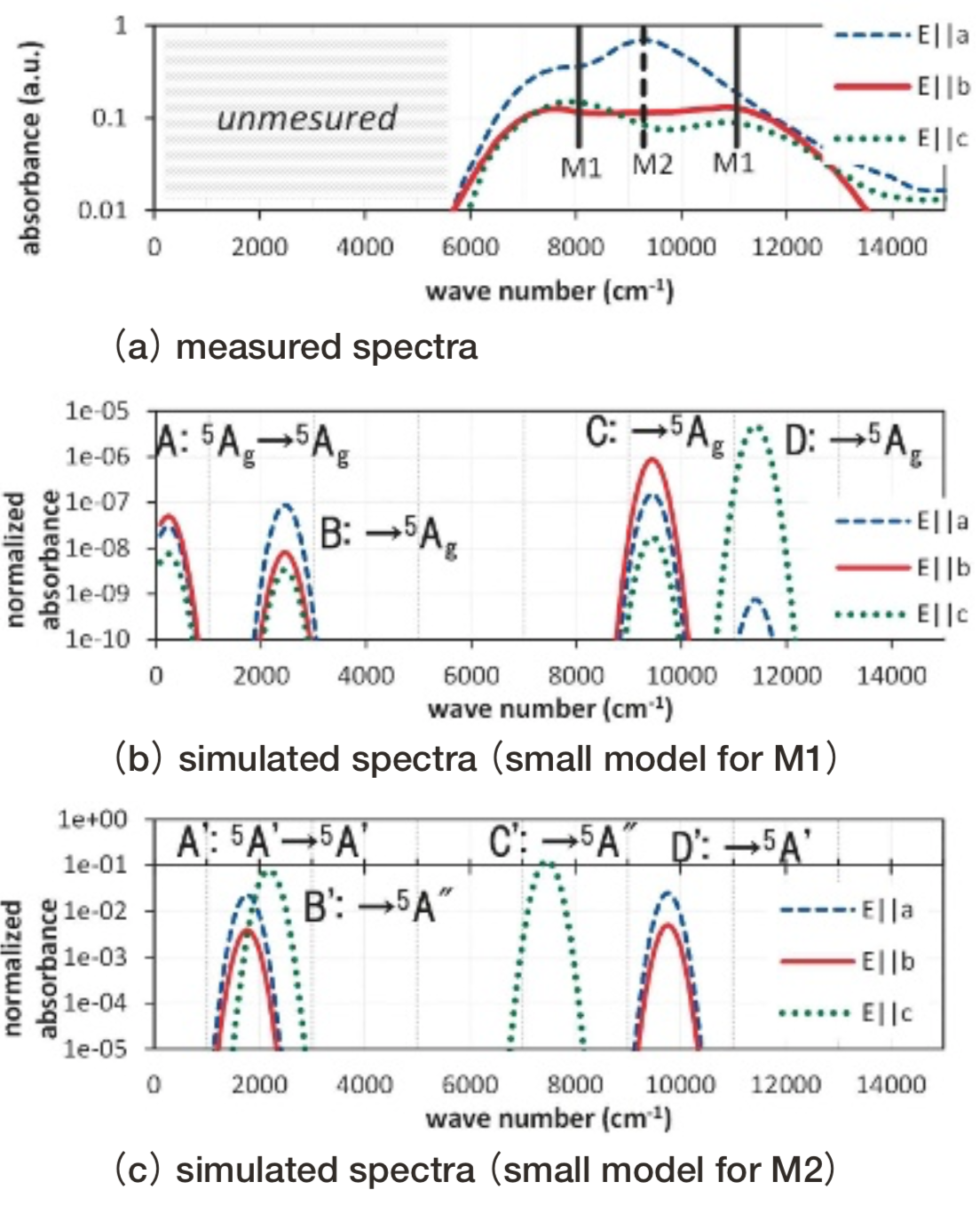 Fig.10 Optical absorption spectra of fayalite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1. Lines in(a): measured spectra of fayalite(17). Vertical bars in(a): peak maxima(18) assigned to M1 and M2 cite respectively.
Fig.10 Optical absorption spectra of fayalite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1. Lines in(a): measured spectra of fayalite(17). Vertical bars in(a): peak maxima(18) assigned to M1 and M2 cite respectively.3.1.3. Siderite
Sideriteの単位胞と小規模モデルをFig. 11に示す。
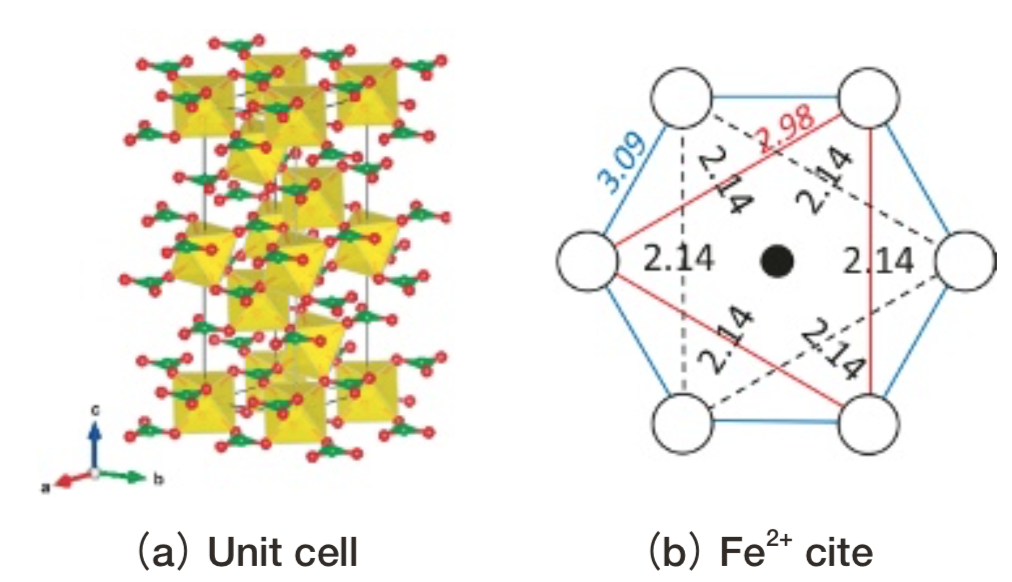 Fig.11 Structure of siderite.(a)shows the unit cell.(b) shows Fe2+ cite projected onto(001). Green triangle: CO3.Yellow polyhedron: FeO6 .(b)shows the small model. ●: Fe2+, ○:O2-. Fe-O or O-O distances are indicated(Å).
Fig.11 Structure of siderite.(a)shows the unit cell.(b) shows Fe2+ cite projected onto(001). Green triangle: CO3.Yellow polyhedron: FeO6 .(b)shows the small model. ●: Fe2+, ○:O2-. Fe-O or O-O distances are indicated(Å).Fig. 12にsideriteの光吸収スペクトルの計算結果を測定データ(17)とともに示す。SideriteはD3h対称で反転対称性を持つため、光吸収強度の計算結果は誤差の範囲で0とみなせるが、測定では振電相互作用により光吸収強度を得ているものと考えられている。測定では約7800, 9500 cm-1の2本の光吸収ピークが観察されているが、計算上は8000~10000 cm-1付近に2重縮退した1つの多重項が存在する。測定の2本のピークの内1本は、エネルギーの近いスピン許容遷移との相互作用により強度を得ているスピン禁制遷移に起因する可能性が指摘されている(12)。今回取り上げた結晶(Table 1)のDV-ME法計算結果にはスピン許容遷移にエネルギーの近いスピン禁制遷移は存在しなかったため、sideriteの測定で見られる2本のピークは動的Jahn-Teller効果で2重縮退が分裂したものと考えられる。
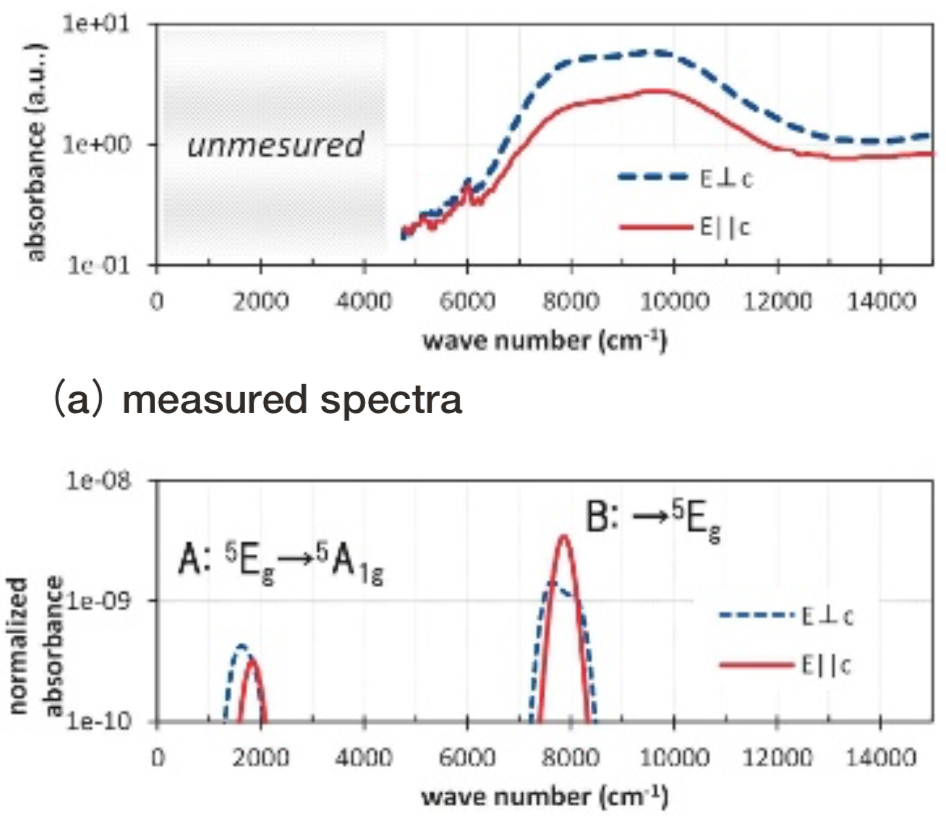 Fig.12 Optical absorption spectra of siderite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1.
Fig.12 Optical absorption spectra of siderite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1.3.1.4. Gillespite
Gillespiteの小規模モデルと大規模モデルをFig. 5に示した。その配位多面体は、Fe2+イオンが面に垂直に僅かに0.03Å飛び出た、正方形に近い構造になっている。Fig. 13にgillespiteの光吸収スペクトル計算結果を測定結果(17)とともに示す。
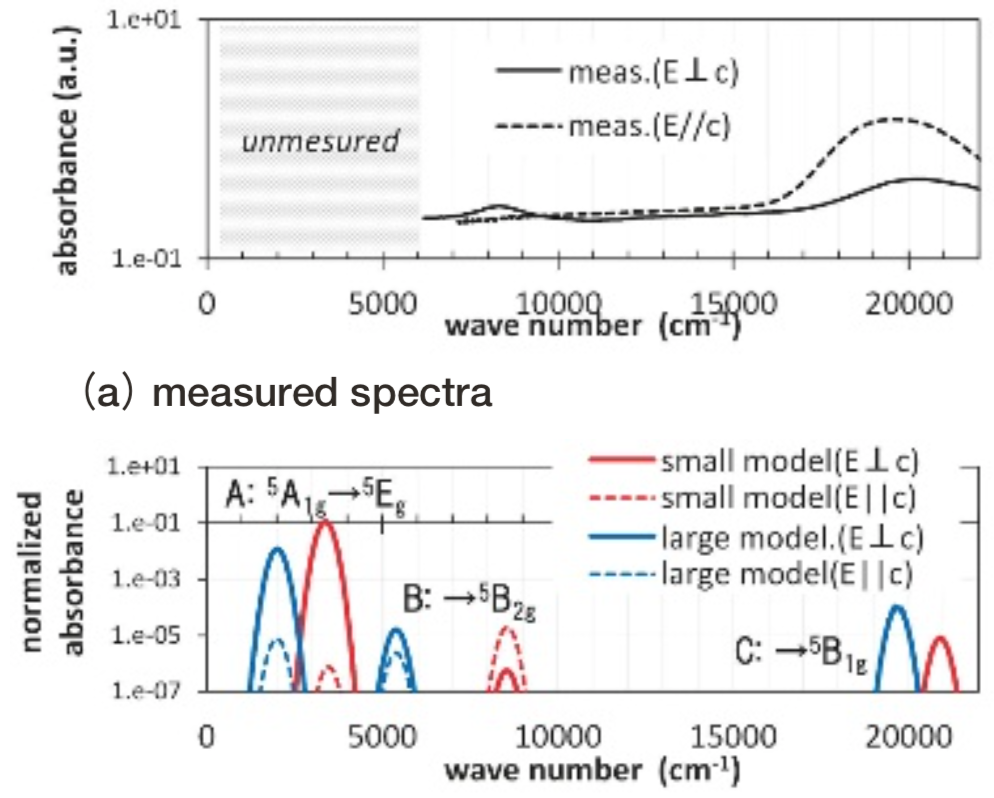 Fig.13 Optical absorption spectra of gillespite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1.
Fig.13 Optical absorption spectra of gillespite. Absorbance is represented in logarithmic scale. Simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1.計算結果のピークBおよびCは、測定の20300, 8400 cm-1付近の光吸収ピークに対応すると考えられる。ピークBおよびCの規格化された光吸収強度の計算値は1e-04未満と僅かである。計算モデルの正確な対称性は反転対称性を持たないC4vであるが、反転対称性を持つD4hとほぼみなせることに整合する。測定では振電相互作用により光吸収強度を得ているものと考えられている。
また、計算上は測定された波数の範囲外である3000 cm-1でピークAが生じているが、異組成類似構造の結晶の測定からgillespiteにおいても3本目の光吸収が2500 cm-1付近に存在すると推測されている(18)。このように計算上得られたピークA, B, Cに関して従来の推察と整合する計算結果が得られている。
3.1.5. その他の結晶
Table 1に挙げた結晶の内、5配位のFe2+イオンを持つgrandidierite(Fig. 4(a)参照)、大きく歪んだ4面体のFe2+イオンを持つpellyiteについては測定データとの定量的な対比は困難であった。Grandidieriteについては複数種のFe2+サイトがあるとされているのに対し1種のサイトの構造データしか入手できなかったため、pellyiteについては測定データにFe2+サイトに計26%置換したMg2+やMn3+等の影響があると考えられるためである。これらgrandidierite, pellyiteの光吸収スペクトル計算結果はFig. 16に掲載する。なお、stauroliteは位置不定なHを含むためクラスターを取り囲む点電荷を考慮しない計算のみ行ったが、測定結果とよく整合する光吸収スペクトルが得られている(16)。Almandineについてはガラス中では存在することが報告されていない8配位であるためここでは紹介しなかったが、測定結果とよく整合する結果が得られている。
3.2. 結晶中Fe2+イオンの光吸収波数の再現性
Fig. 14に、光吸収波数の測定結果との対比が可能であったferrosilite, fayalite, siderite, gillespite, stauroliteについて、測定波数(18)とDV-ME法計算値を対比した結果を示す。ここでとりあげた結晶の計算結果は、3.1章の光吸収スペクトルの計算結果からわかるように、測定波数範囲より低波数側にも基底状態からスピン許容遷移可能な励起状態を持つ。そのため、高波数側の計算値から測定値と対比した。またfayaliteのM2サイトの計算結果は測定波数域に2本のピークC’, D’を持つのに対し、測定ではM2サイトには1本のピークしか帰属されていないという不整合があった(Fig. 10)が、偏光依存性が整合するピークD’で測定値と対比した。
Fig. 14中のferrosilite, fayalite, sideriteおよびgillespiteの 4つの結晶の計算結果からは、配位多面体の周辺イオンを点電荷として考慮した小規模モデルでは、考慮しない場合に比べ光吸収波数の再現性が同等か向上していることが確認できた。配位多面体の周辺イオンを点電荷として考慮した小規模モデルでは、fayaliteの8050 cm-1の測定ピークを1400 cm-1、almandineで1200 cm-1過大評価していた他は、測定との光吸収波数の差は1000 cm-1未満であった。
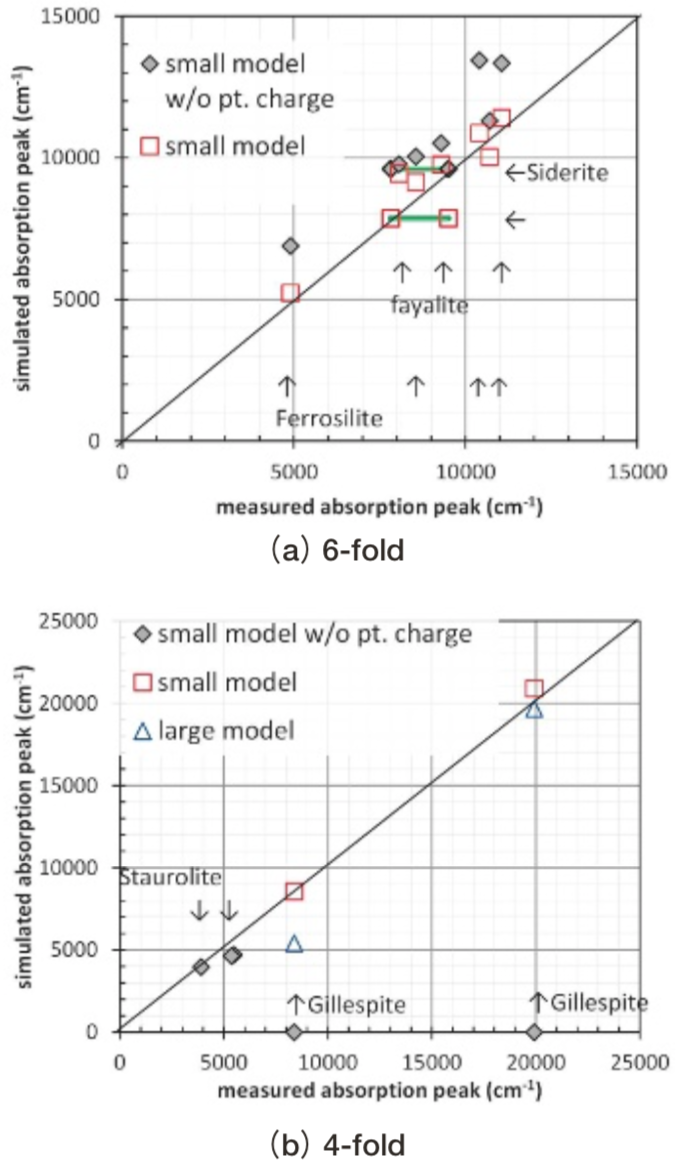 Fig.14 Comparison of measured and simulated wave numbers of absorption attributed to Fe2+ ions in crystals. Solid square: small model without point charge. Open square: small model with point charge. Open triangle: large model with point charge. Two marks connected by green line represent peak split by Jahn-Teller effect.
Fig.14 Comparison of measured and simulated wave numbers of absorption attributed to Fe2+ ions in crystals. Solid square: small model without point charge. Open square: small model with point charge. Open triangle: large model with point charge. Two marks connected by green line represent peak split by Jahn-Teller effect.3.3. SiO2ガラス中Fe2+イオンの光吸収スペクトル計算
SiO2ガラス中に含まれるFe2+イオンの局所構造モデルを作成し光吸収スペクトル計算を行った。モデルの作成には、まず分子動力学法(MD)計算で101原子からなる33SiO2-FeOガラスの構造モデルを作成し、第一原理バンド計算により構造最適化した。密度が2.2 g/cm3になるよう、単位格子形状は一辺11.57 Åの立方体とした。その後DV-ME法計算のためのクラスターモデルを切り出した。33SiO2-FeOはFe2+イオンの濃度が非常に大きい組成であるが、本モデル中でFe2+イオンは互いに11 Å程度離れて存在し、孤立して存在するFe2+イオンの光吸収を評価するための局所構造モデルとしては十分妥当と考えている。
MD計算では、Guillotらによる力場(19)で初期構造はランダムとし平均冷却速度6.7×1013 K/sで7000 Kから室温まで急冷した。第一原理バンド計算では、交換相関相互作用の理論にPBE(GGA)、擬ポテンシャルにVanderbiltのウルトラソフト型、内殻の取り扱いにPAW法を用いた。k点はΓ点のみとし、エネルギー変化および力の収束判定値はそれぞれ2.72×10-7 eVおよび0.01 eV/Åとした。MD計算および第一原理バンド計算には汎用コードであるMXDORTO(20)および PHASE/0(21)を用いた。
MD計算の初期構造を変えることで多様な局所構造が得ることができたが、今回は第一原理計算によるエネルギーが最も低かった構造(Fig. 15(a))のみで光吸収スペクトル計算を行った。その局所構造(Fig. 15(b))をみると、1つのSiO4 4面体と辺共有しており、2つの非架橋酸素、2つの架橋酸素が3.3 Å未満の距離に存在しており、電荷の均衡がとられている。その配位多面体は4面体であるが、gillespite中のFe2+イオン(Fig. 5)のように平面に近い。配位多面体の歪みを表すQ/Eは1.54であり、4配位の Fe2+イオンを含む結晶の中ではQ/Eが1.09と比較的大きいpellyite中のFe2+イオンに近い。
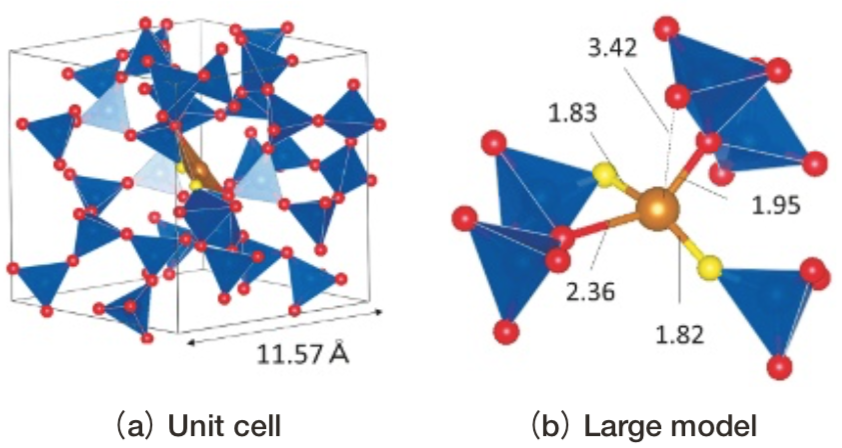 Fig.15 Model of SiO2 glass including Fe2+ ion.
Brown ball: Fe2+. Red ball: bonded oxygen ion. Yellow ball: non-bonded oxygen ion. Blue polyhedron: SiO4 with r(Si-O)<2.0Å .Brown polyhedron: FeO4 with r(Fe-O)<3.3 Å.
Fig.15 Model of SiO2 glass including Fe2+ ion.
Brown ball: Fe2+. Red ball: bonded oxygen ion. Yellow ball: non-bonded oxygen ion. Blue polyhedron: SiO4 with r(Si-O)<2.0Å .Brown polyhedron: FeO4 with r(Fe-O)<3.3 Å.Fig. 16(a)に、ガラス構造モデル(Fig. 15)中Fe2+イオンについて、小規模モデルあるいは大規模モデルの2通りで計算した光吸収スペクトル計算を示す。Fig. 16(b), (c), (d)にはそれぞれ、今回のガラス構造モデルと比較的似ているgillespiteおよび pellyiteと、 5配位および歪んだ6配位の代表としてgrandidierite(Fig. 17(d))およびferrosiliteのM2サイト(Fig. 7(c))の小規模モデルによる光吸収スペクトルを示す。
Fig. 17にガラスや結晶中のFe2+イオンのd軌道を主成分とする分子軌道の形状の計算結果を、各構造モデル中でのエネルギーの序列とともに示す。配位子(O2-イオン)の原子軌道とも混成していることが分かる。当然ではあるが最低エネルギーの分子軌道は配位子が少ない方向に広がり、最高エネルギーの分子軌道は配位子の影響を受ける方向に広がっている。中間のエネルギーを持つ分子軌道の広がる方向や形状は各結晶で異なる。分子軌道の分裂は、Fe-O距離の影響や配位子との位置関係を敏感に受けると考えられる。
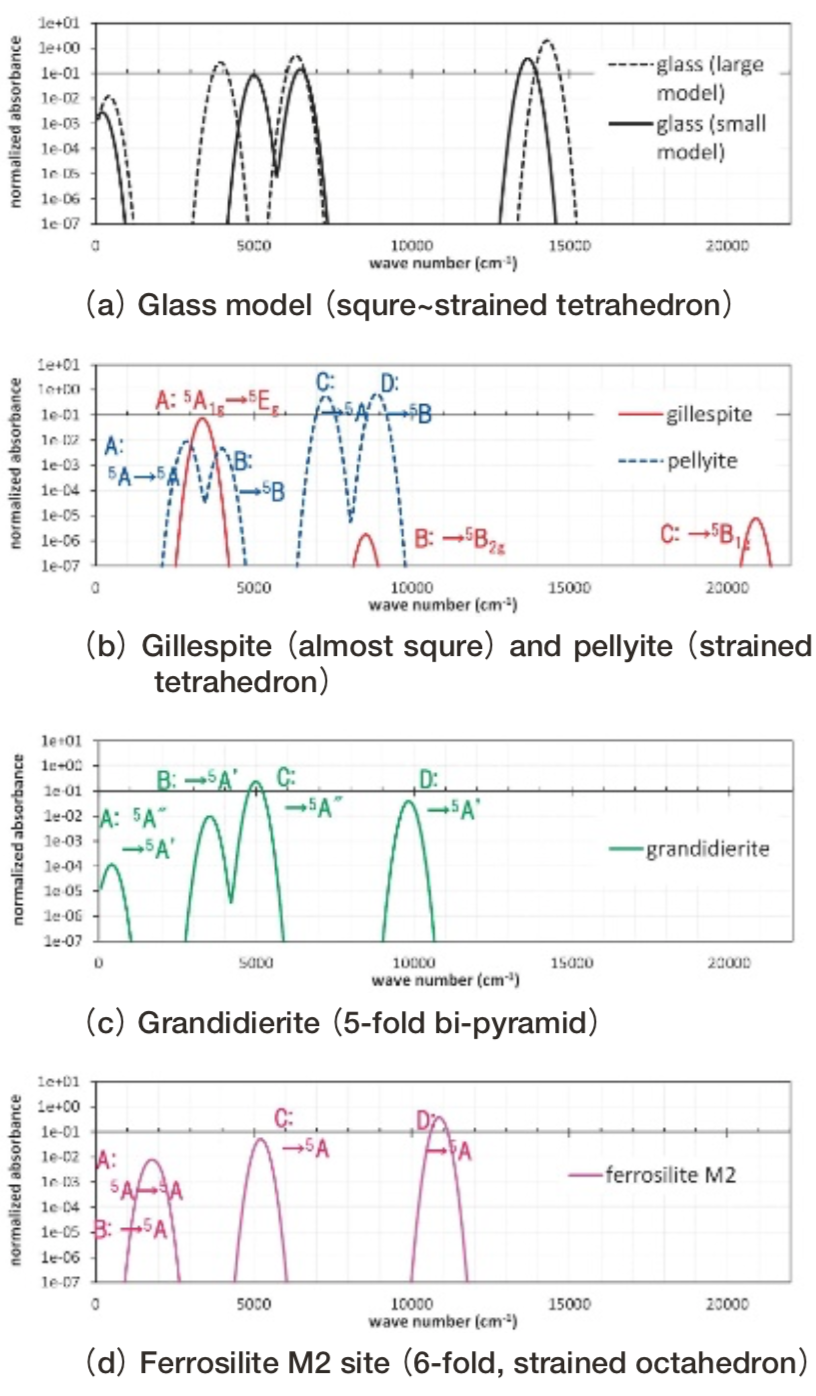 Fig.16 Simulated optical absorption spectra attributed to Fe2+ ion in the glass model, 4-fold , 5-fold and 6-fold crystals. Three polarized spectra are averaged. The simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1.
Fig.16 Simulated optical absorption spectra attributed to Fe2+ ion in the glass model, 4-fold , 5-fold and 6-fold crystals. Three polarized spectra are averaged. The simulated absorbance is normalized so that the simulated value of Fe2+ ion in spinel Mg site using small model turns to be 1.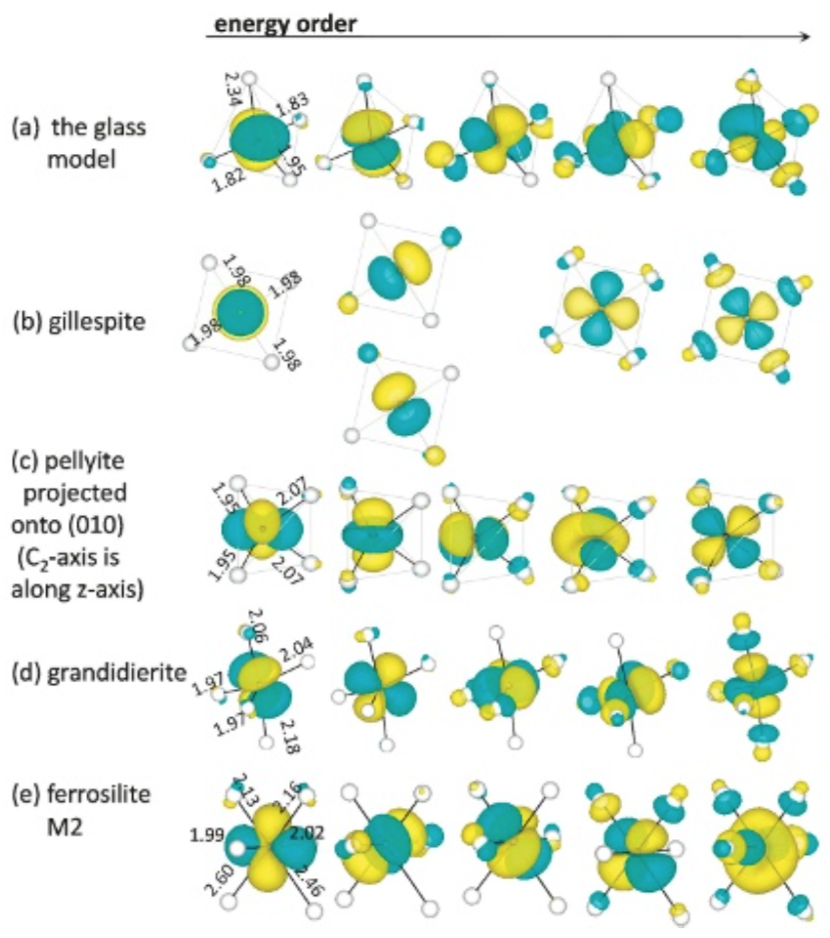 Fig.17 Molecular orbitals consisting mainly from d-orbitals. From top to bottom, the case of the glass model, gillespite, pellyite, grandidierite and ferrosilite(M2 cite).
Fig.17 Molecular orbitals consisting mainly from d-orbitals. From top to bottom, the case of the glass model, gillespite, pellyite, grandidierite and ferrosilite(M2 cite).4. 考察
4.1. 光吸収波数の計算精度
DV-ME法計算により結晶中のFe2+イオンのクラスターモデルを用いて光吸収スペクトルを評価した。従来のLF理論で正確に扱うことの困難な対称性の低い局所構造のFe2+イオンを持つ結晶においても、配位多面体の周辺イオンを点電荷として考慮することで主要な偏光成分や光吸収波数について、測定結果とおおむね整合する光吸収スペクトルを計算できている。一方でfayaliteの場合だけは測定データとの整合性がやや乏しい結果となった。整合性が乏しい要因としては、Feイオンの配位多面体が3次元的につらなっているため、隣り合うFeイオンのd軌道との混成が強く、現在のクラスターサイズのモデルでは配置間相互作用を精度よく求めるのが困難なためと推測している。
実績対比の可能であった結晶ferrosilite, fayalite, siderite, gillespite, almandineでは、周辺イオンを点電荷として考慮した小規模モデルによるピーク波数計算値は実測値を-700~1400 cm-1未満の差で再現した。DV-ME法は対称性によらずに一貫した理論に基づく解析ができるため、ガラス中で想定されるような歪んだ局所構造であっても同程度の計算精度が期待できる。
一方、-700~1400 cm-1の計算精度では、ガラス組成の調整に対するFe2+イオンの光吸収スペクトルの変化(ピーク値にして数百cm-1程度)を議論する目的には不十分である。DV-ME法計算においてはルビーでその効果が報告されている配置依存補正や相関補正(13)(14)が計算精度の向上に有用と思われる。また、DV-ME法に限らず第一原理計算の枠組みで様々な近似手法あるいは高精度手法が開発されており、今後の発展が期待される。
4.2. 配位多面体の周辺イオンの影響
GillespiteおよびSiO2ガラス構造モデルについては、小規模モデルと大規模モデルそれぞれで光吸収スペクトル計算を行った。大規模モデルの小規模モデルからの差は、gillespite(Fig. 13)およびガラス(Fig. 16)の場合、光吸収強度についてはそれぞれ0.1~10倍および10倍程度、光吸収波数については-1200~3000 cm-1および-1000~1000 cm-1程度の差がある。大規模モデルと小規模モデルとで、光吸収波数の評価は0.1~10倍、吸収強度の評価は数千 cm-1程度変わる可能性がある。
原理的には大規模モデルの方が高精度であるが、gillespiteに関しては小規模モデルの方が測定の光吸収波数を再現している。小規模モデルで周辺イオンとの共有結合を考慮していないことに起因する計算誤差が、電子状態の計算誤差とキャンセルした可能性がある。
4.3. 局所構造と光吸収スペクトルの関係
SiO2ガラス構造モデル中のFe2+イオンについて、DV-ME法計算により光吸収スペクトルを評価し、局所構造との関係の考察を試みた。
今回作成したガラス構造モデル中のFe2+イオンは4配位で、配位子場の対称性が低いためd軌道を主成分とする分子軌道は縮退が解けて5つに分裂し、DV-ME法計算によると14000 cm-1に比較的大きい光吸収を示す。結晶中の4配位の場合では、2.7~3.4 Å程度離れたFe3+イオンへの電荷移動遷移 (18)を別にして、14000 cm-1付近に比較的大きい光吸収を示す結晶は筆者の知る限りは見つかっていない。6配位ではtourmalineのAlサイトに置換したFe2+イオンはFe-O距離が1.93Åと短いため大きい配位子場効果が働き、14500 cm-1に光吸収を持つ。以上のことから、光吸収波数域の情報のみから4配位に帰属することが困難であることがわかる。
今回作成したガラス中のFe2+イオンと、配位多面体形状が比較的似ているgillespiteやpellyite中のFe2+イオンとの間では、光吸収スペクトルおよび分子軌道に、局所構造の類似性を示すような共通点を見出せなかった。光吸収スペクトル形状に関しては比較的grandidieriteがガラス構造モデルの計算結果に類似している(Fig. 16)が、対応する状態の分子軌道形状に類似性は見出せない(Fig. 17)ため、光吸収スペクトルが類似していたのは偶然と考えられる。
以上のことから、構造が既知の結晶中のFe2+イオンの光吸収スペクトル形状による指紋照合の方法では、対称性の不明な局所構造を持つFe2+イオンによる光吸収スペクトルから局所構造へ帰属することは原理的に困難であるとわかる。ガラス中のFe2+イオン周辺の局所構造と光吸収スペクトルの関係解明のためには、対称性や配位数など局所構造の情報を事前にできるだけ絞り込むことが重要である。
4.4. Fe2+イオン周辺の局所構造の存在割合
ガラス中のFe2+イオンについて想定した局所構造モデルの存在割合を、そのDV-ME法計算結果と測定の光吸収スペクトルの比較により推測することができる。たとえば14000 cm-1付近光吸収は、ソーダライムガラス中Fe2+イオンの測定結果では他の吸収ピークに比べ相対的に弱い(Fig. 1)。一方、今回のガラス構造モデルでは14000 cm-1付近の規格化された光吸収強度の計算結果は0.1~1と比較的大きい。すなわち、本計算で想定したような局所構造および周辺電荷分布を持つFe2+イオンが実際のソーダライムガラス中に存在するとしても、割合は僅かであることが推定できる。さらに正確に存在割合を議論するためには、光吸収波数や光吸収強度の計算精度を向上する必要がある。熱振動によるスペクトルの拡幅の考慮、ガラス中Fe2+イオン周辺の局所構造の存在割合も含めたモデル化、ガラス修飾イオンの影響の把握、また今回の計算結果のSiO2ガラス中Fe2+イオンの光吸収スペクトル測定データとの対比も今後の課題である。
5. おわりに
ガラス中に含まれるFe2+イオン周辺の局所構造と光吸収スペクトルの関係の解明に向けた、第一原理計算の一種であるDV-ME法による取り組みについて紹介した。
Fe2+イオンの配位多面体とその周辺イオン位置に置かれた点電荷から構成される局所構造を適切にモデル化することにより、最大で1400 cm-1程度の誤差で、局所構造の対称性を問わずに一貫した理論に基づき光吸収波数を再現できることを、5結晶中の計7個の局所構造モデルにおいて確認した。また、光吸収強度に関しても測定結果とおおむね整合する結果を得た。その結果、ガラス中で想定される歪んだ局所構造モデルによる光吸収スペクトルを評価することで、その局所構造モデルがガラス中で存在しうるかを定性的に評価できるようになった。対称性の低い局所構造に関するこうした評価は、経験的パラメータと高い対称性を仮定する必要がある配位子場理論に基づく解析では困難である。また、ガラス中で予想されるFe2+イオン周辺の大きく歪んだ局所構造と光吸収スペクトルの関係を、Fe2+イオンのd軌道からなる分子軌道の観点から、解釈することが可能となった。
しかしながら、ガラス組成とガラス中のFe2+イオン周辺の局所構造の関係を予測する技術はまだ構築されていないため、ガラス組成、ガラス中Feイオン周辺の局所構造、光吸収スペクトルの三者を総合的に予測できる技術構築は、まだ道半ばである。
今後、さらに光吸収スペクトルの計算精度を向上させ、正確なガラス構造の予測技術を確立することにより、Feイオンによる光吸収を制御したガラス組成設計へ貢献していきたい。
6. 謝辞
パリ大学のProf. Calas, Dr. Lelong, Dr. Vercamerらには、RIXS等の測定および多重項計算など多角的な解析および大変有益な議論をしていただいた。ここに謝意を表する。
DV-ME法の利用に関しては、開発者である関西学院大学の小笠原一禎教授にご指導いただいた。ここに謝意を表する。
参考文献
- 山根正之ほか 編, ガラス工学ハンドブック, (朝倉書店, 1999)
- Y. Kondo, et al., 23th International Congress on Glass (2013).
- 特開2016-84281
- 上村洸ほか, 配位子場理論とその応用, (裳華房, 1969)
- 近藤裕己, 土屋博之, SPring-8利用報告書, 課題番号2010B3350, (2010).
- 近藤裕己, 土屋博之, SPring-8利用報告書, 課題番号2010B3236, (2010).
- 岡島敏浩ほか, NEW GLASS, 27, 8,(2012).
- V. Vercamer, Ph.D. thesis, Universit´e Pierre et Marie Curie, 2015.
- T. T. Volotinen, J. M. Parker, and P. A. Bingham, Phys. Chem. Glasses, 49, 258, (2008).
- M. A. Arrio, et al., Europhys.Lett. 51, 454, (2000).
- M. Hunault, et al., J. Phys.: Conf. Ser. 712, 012005, (2016).
- . Vercamer, et al., Phys. Rev. B 94, 245115, (2016)
- K. Ogasawara, et al., Phys. Rev. B 61, 143, (2000).
- S. Watanabe, et al., Phys. Rev. B 79, 075109, (2009).
- 小和田善之ほか, 足立裕彦 監修, はじめての電子状態計算~DV-Xα法分子軌道計算への入門~, (三共出版株式会社, 1998).
- 藤田早苗, 高田章, 小笠原一禎, DV-Xα研究協会会報, 28, 162,(2015).
- G. R. Rossman’s website, http://minerals.gps.caltech.edu/.
- R. G. Burns, Mineralogical Applications of Crystal Field Theory, (Cambridge University Press, 1993).
- B. Guillot, and N. Sator, Geochimica et Cosmochimica Acta, 71, 1249, (2007).
- 平尾一之, 河村雄行, パソコンによる材料設計, (裳華房, 1994).
- T.Yamamoto, et al., Phys. Lett. A 373, 3989, (2009)