AGC Research Report 73(2023)
Improving Deep Neural Network in Predicting Electron Ionization Mass Spectra with Molecular Similarity-wise Sampling
分子類似度サンプリングによる深層ニューラルネットワーク 電子イオン化マススペクトル予測の改善
山口凌平*・竹田薫識*・山田正嗣*・柿内俊文*・今村 穣*
Ryohei Yamaguchi, Shigenori Takeda, Masatsugu Yamada, Toshifumi Kakiuchi, and Yutaka Imamura
*AGC株式会社 先端基盤研究所(ryohei.yamaguchi@agc.com)
質量分析は、有機合成やメタボロミクスなど、さまざまな用途で分子を同定するために広く使用されている手法である。最近、マススペクトルを予測するディープニューラルネットワークモデルが開発され、その高い精度が定量的に評価された。しかし、このモデルは高フッ素化化合物などの特定のターゲットに対しては予測精度が低いことが確認された。そこで、本研究では、分子類似性を利用したデータセットのアンダーサンプリングスキームを導入した。アンダーサンプリングされたデータセットで訓練されたモデルは、フッ素化合物に対する予測性能が向上し、かつ、フッ素化されていない化合物に対する精度も比較的維持されることがわかった。この精度向上は、分子フィンガープリントのビットコリジョンが減少したことに起因すると考えられる。
Mass spectrometry is a technique extensively utilized in various fields, including organic synthesis and metabolomics, to identify molecules. Recently, a deep neural network model was developed to predict mass spectra, and its effectiveness was evaluated. However, we found that the model was unable to identify certain unknown molecules, such as highly fluorinated compounds. To enhance the predictability for these minor compounds, we implemented a dataset undersampling method based on molecular similarity. The model trained using this undersampled dataset showed improved predictability for fluorinated compounds, without compromising the accuracy of identifying non-fluorinated compounds. This improvement in performance could be attributed to the reduction of hash collisions of molecular fingerprints.
1. 緒言
質量分析は低分子化合物の同定に欠かせない技術の1つである。これまで、有機合成物の特性評価やメタボロミクス、環境分析など、さまざまな用途で化合物の同定に用いられている。質量分析はガスクロマトグラフィーと組み合わせられて(GC/MS)、頻繁に使用される。GC/MSは70eVの電子イオン化(EI)で標準化され、30年以上に渡ってスペクトルデータが蓄積されてきた[1,2]。しかし、1億種以上の既知化合物のケミカルスペースと比べるとデータベースは依然小さく、未知スペクトルが多く検出される[3]。実験データベースの網羅率が限られているという現状を克服するために、計算科学を用いて分子構造からスペクトルを推定し、データベースを拡張する方法が用いられる。マススペクトルを計算するために、量子力学に基づく方法[4–9]や機械学習[10,11]による方法が提案されてきた。
Quantum-Chemical electron ionization massspectra(QCEIMS)[4]は第一原理分子動力学と統計解析を組み合わせた方法で、データベースや経験的なフラグメンテーションのルールがなくとも、自動でマススペクトルを予測することが可能だが、高い計算コストを必要とする[12]。一方、EI-MS予測用の深層ニューラルネットワークモデルであるNeural electronionizationmass spectrometry(NEIMS)[10]は100万分子の予測を約90分で実行することが可能である。NEIMSでは、residual networks(ResNets)[13]にbidirectional architectureと呼ばれる質量が高いマススペクトルピークを精度良く記述できるスキームを組み込んでおり、予測性能も従来の機械学習モデルより優れている。
本研究では、フッ素含有率が高い化合物に対するNEIMSの予測性能を評価した。数値評価に基づいて、分子類似度サンプリング(Molecular similarity-wisesampling; MSS)アルゴリズム(Fig.1)を使用したアンダーサンプリングアルゴリズムを提案する。提案された方法は分子フィンガープリントであるExtended-connectivity fingerprints(ECFPs)[14]で見積もった分子類似度を用いている。
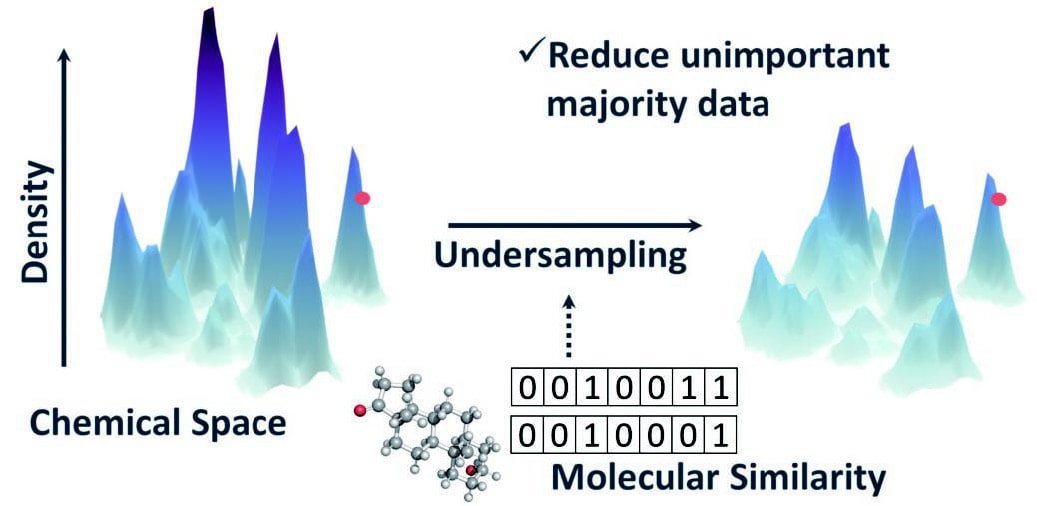 Fig.1 : Undersampling using molecular similarity. Thinning out majority data from clusters that differ from the target. The red circle indicates the position of target molecules.
Fig.1 : Undersampling using molecular similarity. Thinning out majority data from clusters that differ from the target. The red circle indicates the position of target molecules.2. 方法
本研究では、分子構造とEI-MSスペクトルの相関を学習した回帰モデルを用いてマススペクトルの予測を行った。回帰モデルにはNEIMS(Fig.2)を用いた。NEIMSの詳細なアーキテクチャは先行研究[10]に記載されている。実装の仕方はGitHubに公開されている。NEIMSでは、分子構造の特徴量として分子のトポロジー情報を効果的に表現する手法であるECFPs[14]が用いられている。ケモインフォマティクスツールRDKit[15]を使用して、標準のバイナリフィンガープリントでは無く、カウントタイプのECFPsをradius=2で計算した。ECFPsはハッシュ化によって長さ4096の固定長のベクトルとして、ResNetsのインプットとした。ResNetsのアウトプットから直接線形層でスペクトルを予測する方法はforward predictionと呼ばれる。入力のECFPsは分子の局所的な構造を捉えたものであるため、一般に小さな部分構造のピークをより正確に捉えることができる。一方で、forward predictionでは大きなフラグメントのピーク強度を正確に捉えることが難しい。そこで、小さなフラグメントが分子から脱離することを仮定し、forward predictionのモデルを利用しながら大きなフラグメントのピークを予測するreverse predictionを導入した。これらのforward predictionとreverse predictionを利用し、最終的な予測値bidirectional predictionを得ている[10]。
 Fig.2 : NEIMS pipeline.
Fig.2 : NEIMS pipeline.データセットとサンプリング
本研究では、スペクトル予測モデルの学習と評価にNIST 2017 Mass Spectral Library(NIST17)[16]のスペクトルデータを用いた。NIST17 はmainライブラリとreplicatesライブラリで構成される。mainライブラリは267376種のマススペクトルデータから成り、replicatesライブラリは、mainライブラリにも含まれる約23000種の化合物のマススペクトルから成る。replicatesライブラリのスペクトルは、mainライブラリよりもよりノイズを含んでおり、より実験でのばらつきを有するスペクトルに対応していると考えられる。そのため、今後の実用的な活用を見据え、予測モデルのパフォーマンス評価にreplicatesライブラリを使用した。これらreplicatesライブラリに含まれる化合物のスペクトルデータをmainライブラリから除き、NEIMSモデルに学習させた。
スペクトル予測タスクでは、高フッ素含有率化合物のようなマイナー化合物の予測精度が低くなる傾向がある。この要因はNEIMSのデータ表現に使われるECFPsがビットコリジョンという表現力の課題を抱えていること(Fig.3)[17]とデータの不均衡性にあると考えられる。データの不均衡問題に対して、最もストレートでよく使われるアプローチがオーバーサンプリングやアンダーサンプリングといったサンプリング手法である[18–23]。しかし、サンプリング手法の多くはラベルありのクラス分類タスク用に設計されており、マススペクトル予測のような回帰タスクに直接応用することはできない。
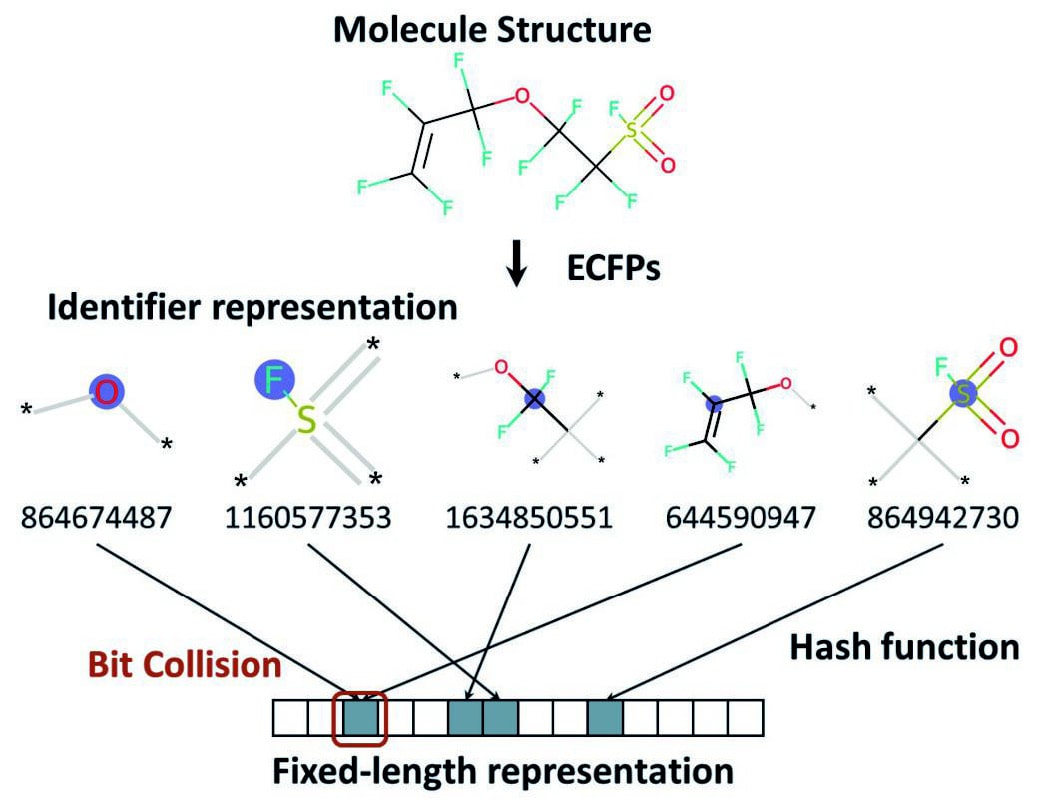 Fig.3 : The examples of identifiers from ECFPs and bit collision. An identifier representation refers to a unique identifier that is assigned to a specific chemical structure pattern. Bit collision refers to the phenomenon in which different data inputs are assigned to an identical hash value in a hash function.
Fig.3 : The examples of identifiers from ECFPs and bit collision. An identifier representation refers to a unique identifier that is assigned to a specific chemical structure pattern. Bit collision refers to the phenomenon in which different data inputs are assigned to an identical hash value in a hash function.本研究では、任意の複数のターゲット化合物の存在を想定して、それらとの分子類似度を全データセットについて計算し、閾値によってデータのラベル付けを行い、類似度の低いデータをアンダーサンプリングした。使用した分子類似度Tanimotoインデックスは次式で与えられる:
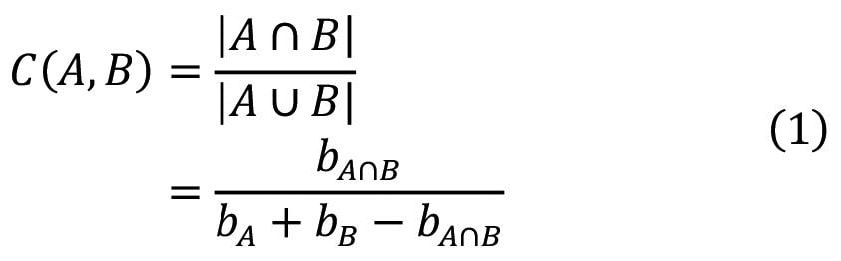
ここでAとBは2つの分子のフィンガープリントの集合であり、bA、bB、bA∩Bは集合A、B、A∩Bのフィンガープリントの数である。
ターゲット化合物j∈[1,n]との類似度が小さい化合物にラベル付けするために、フィンガープリントの集合Ajを用いて、学習データの化合物iのラベルLiを次の通り定義した:
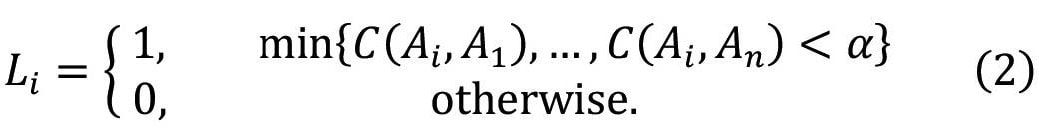
ここではTanimotoインデックスに与える非類似判定の閾値である。
本研究では、任意のフッ素化合物5種(Fig.4)をターゲット化合物としてサンプリング手法のパフォーマンスを評価した。
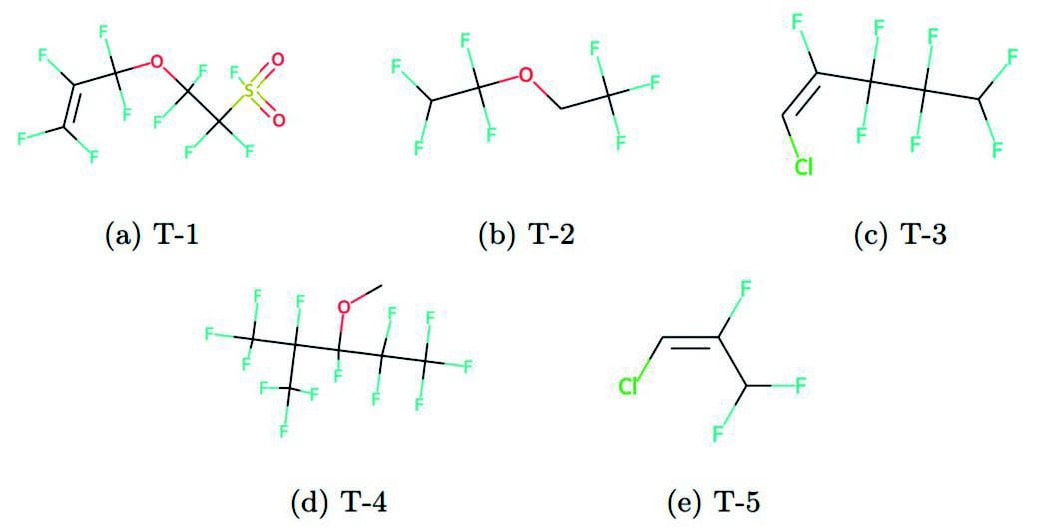 Fig.4 : Target fluorine compounds used for undersampling.
Fig.4 : Target fluorine compounds used for undersampling.(a)1, 1, 2, 2-Tetrafluoro-2-(pentafluoroallyloxy) ethanesulfonyl fluoride,(b)1, 1, 2, 2-Tetrafluoroethyl 2, 2, 2-trifluoroethyl ether,(c)(E)-1-chlor o-2, 3, 3, 4, 4, 5, 5-heptafluoropent-1-ene,(d)Pentane, 1, 1, 1, 2, 2, 3, 4, 5, 5, 5-decafluoro-3-methoxy4-(trifluoromethyl)-,(e)(E)-1-Chloro-2, 3, 3-trifluoro-1propene
予測スペクトルの評価
予測スペクトルを評価するために、ライブラリに登録されている既知スペクトルと比較し予測精度を評価する必要がある。一般的にスペクトル間の類似性を見積もる指標を用いて比較を行うが、適切な指標の選択が重要である[24,25]。質量分析ソフトウェアでは、加重コサイン類似度が一般的に使用されている。加重コサイン類似度は以下の式で表される[24]:
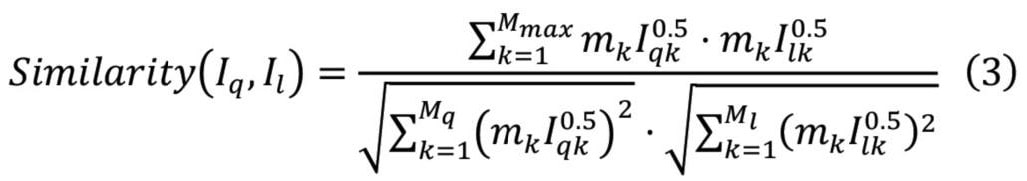
ここでzとmは電荷とフラグメントイオンの質量であり、kはm/zのインデックスを表す。IqkとIlkはそれぞれ検索対象となるクエリスペクトルと参照されるライブラリスペクトルの強度であり、MqとMlはIqとIlが非ゼロの最大のインデックス、MmaxはMqとMlの最大値を表す。IqkとIlkが0以上の場合、加重コサイン類似度は、0から1の範囲で値を取る。値が1に近いほどスペクトルは類似しており、値が0に近いほどスペクトル同士の無相関を示す。本研究では、予測スペクトルと観測スペクトルの加重コサイン類似度を機械学習モデルのパフォーマンス評価に用いた。
スペクトル類似度の評価に加えて、スペクトルライブラリのマッチング性能を評価するために、観測スペクトルとモデルに予測されたスペクトルから構成されたライブラリを用いた。具体的には、NISTメインライブラリからクエリ分子に対応するスペクトルを削除し、予測スペクトルに置き換えて、クエリスペクトルと拡張ライブラリのスペクトル間の類似度を計算した(Fig.5)。クエリスペクトルに対応した、スペクトルの類似度のランクでライブラリマッチング性能を評価した。
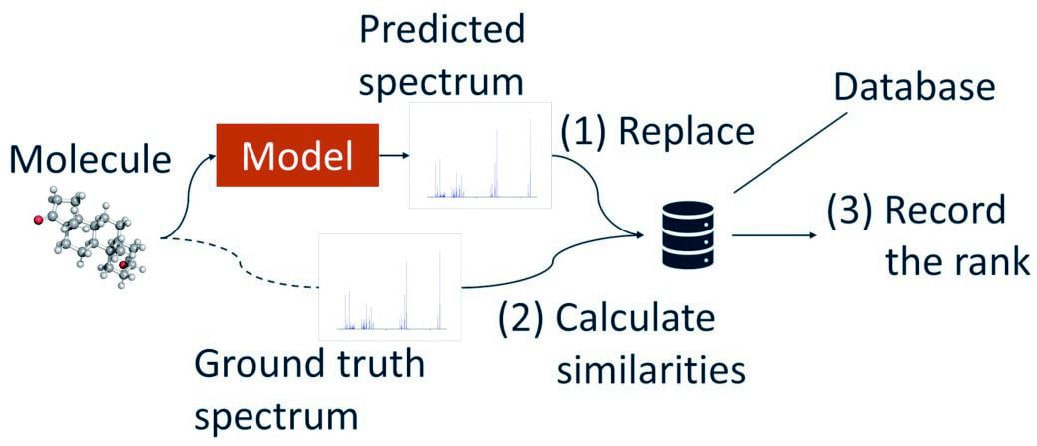 Fig.5 : Library matching rank score.(1)Replace the relevant spectrum data in the database with predi cted spectrum.(2)Search the database with correct ground truth spectrum and sort each data by similarity.(3)Rank the replaced predicted spectrum as scores.
Fig.5 : Library matching rank score.(1)Replace the relevant spectrum data in the database with predi cted spectrum.(2)Search the database with correct ground truth spectrum and sort each data by similarity.(3)Rank the replaced predicted spectrum as scores.3. 結果と考察
分子類似度を利用したアンダーサンプリング手法の効果を評価するために、NIST17 main ライブラリの236715個のスペクトルデータを利用した。式2のαを0.1として、Li=1のデータを70%アンダーサンプリングした。アンダーサンプリング後のデータセットと同じサイズになるようにランダムサンプリングしたデータセットを比較に用いた。
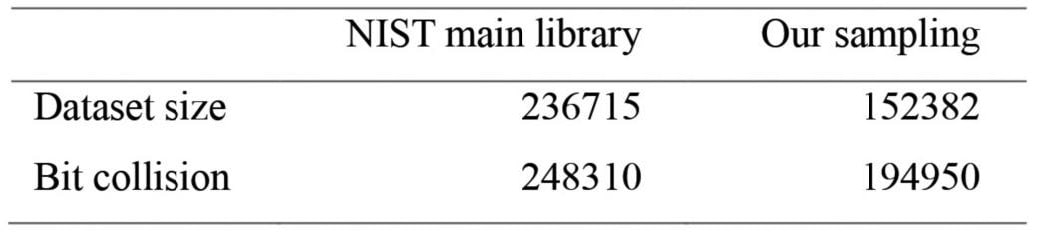
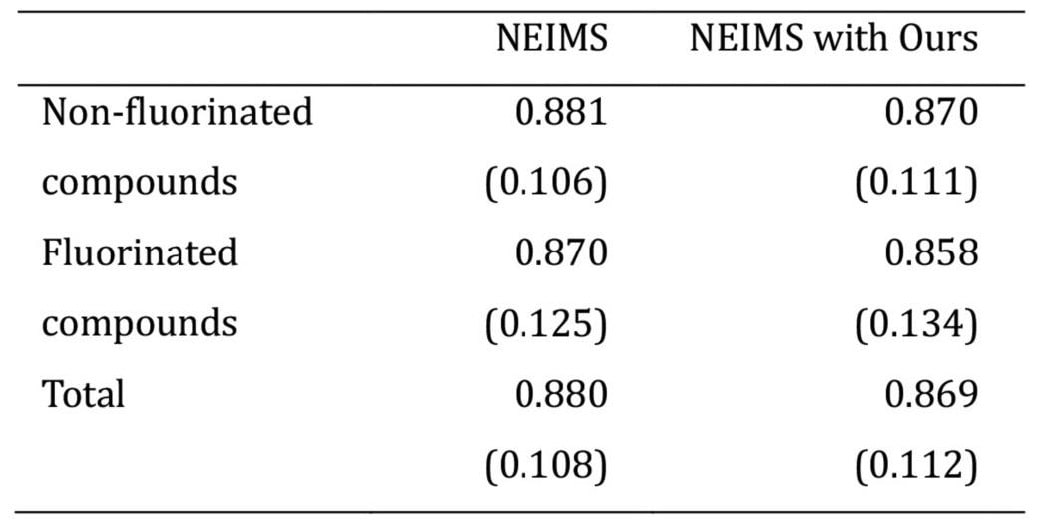
サンプリング手法の評価に先立ち、各種データセットについてECFPsを計算し、ビットコリジョンが発生しているidentifier(部分構造に付与される固有の識別子)の数を調べた(Table 1)。NIST main ライブラリの中で学習に用いたデータセットにおいて約25万個もビットコリジョンしていることが明らかになった。一方、我々の提案するアンダーサンプリングによってビットコリジョンは約21%削減された。
サンプリング前後のデータセットのケミカルスペースを次元圧縮手法UMAP[26]で可視化した(Fig.6a)。分子構造はmol2vec[27]で数値ベクトル化を行った。mol2vecはECFPsをベースとした教師なしの分子表現学習手法であり、ECFPsはNEIMSの入力データとして使用されている。Fig.6aの曲面はカーネル密度推定法で推定された確率密度をデータセットサイズで規格化している。高密度の領域から多くのデータが除外されたことが確認された。また、Figs. 6b、6cに示すように、ターゲット化合物と非類似の化合物が除外され、ビットコリジョンが緩和された。
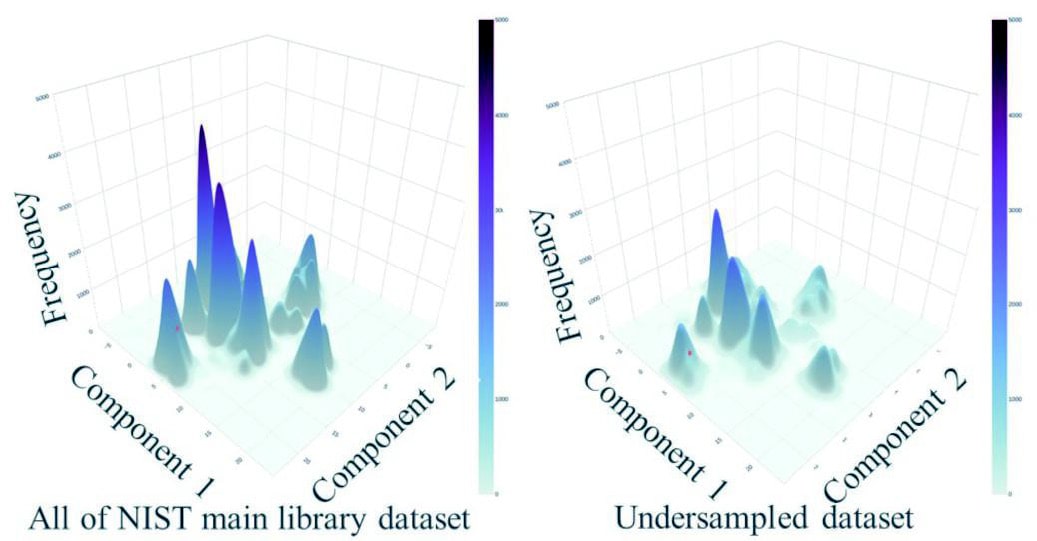 (a)Kernel density estimation surface
(a)Kernel density estimation surface (b)The examples of dropped compounds.
(b)The examples of dropped compounds. (c)The bit collision fingerprint examples.Fig.6 : Results of the sampling process, showing the distri bution of data and examples of data points excluded by the sampling method.
(c)The bit collision fingerprint examples.Fig.6 : Results of the sampling process, showing the distri bution of data and examples of data points excluded by the sampling method.EI-MS スペクトル予測パフォーマンス評価
最初に、サンプリング手法のパフォーマンスを、機械学習モデルの訓練時にvalidationに使用しなかったNIST replicatesライブラリ中のスペクトルデータ11600点を用いて評価した。Table 2に見られるように、サンプリングされたデータによって学習されたモデルでは、加重コサイン類似度が通常の全データで学習されたモデルに対して平均で0.011減少した。これはわずかな差で実用上大きな差ではないと考えられる。
次に、サンプリング手法がターゲット化合物として使用された化合物の予測パフォーマンスに与える影響を調べた。観測スペクトルと予測スペクトルの加重コサイン類似度とライブラリ検索順位の2つのスコアを指標として、NIST main ライブラリ全データを使用した場合(以下、全データモデル)とアンダーサンプリングを実施した場合(以下、アンダーサンプリングモデル)をFig.7に比較する。
加重コサイン類似度は全ての化合物に対して向上し、最大では0.265も増加した。全データモデルの予測類似度が低い化合物ほど、サンプリングによる効果が大きいことがわかる。ライブラリ検索順位を見ると、全データモデルで既に1位だったT-5を除いて順位が改善された。
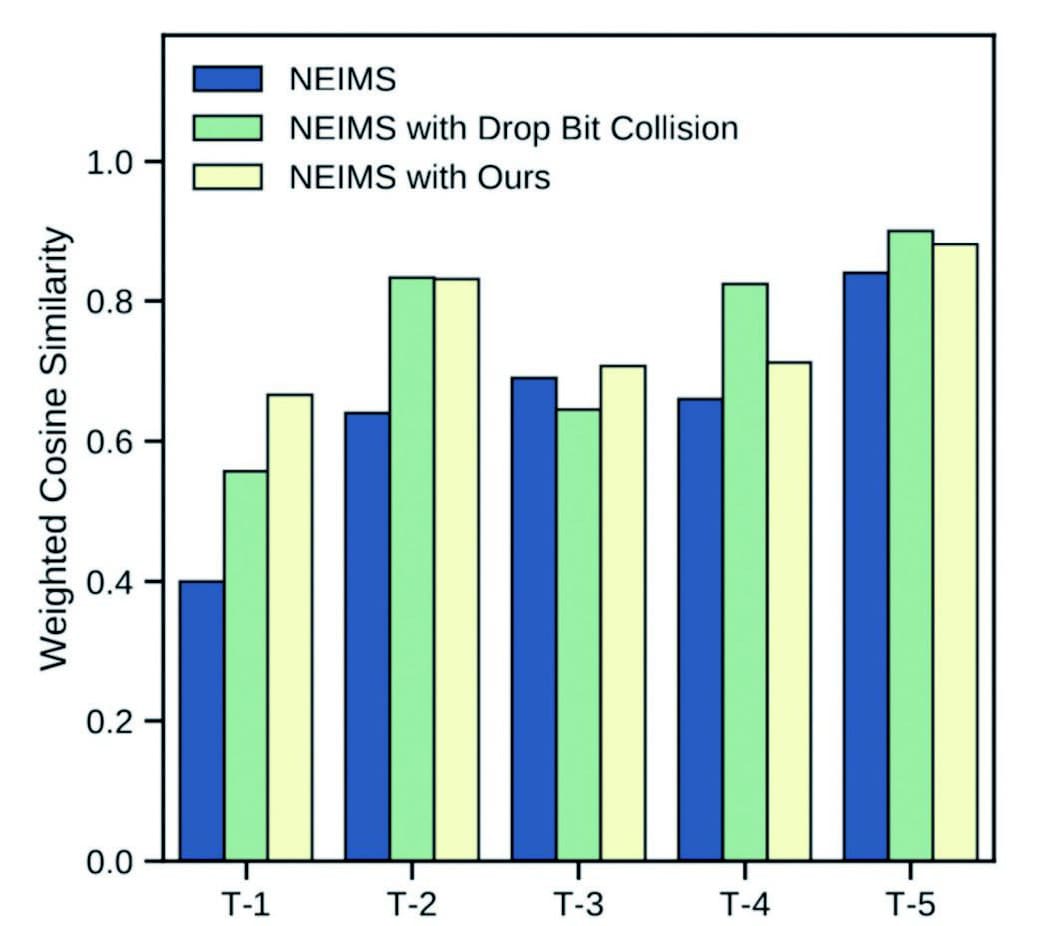 (a)Weighted cosine similarity
(a)Weighted cosine similarity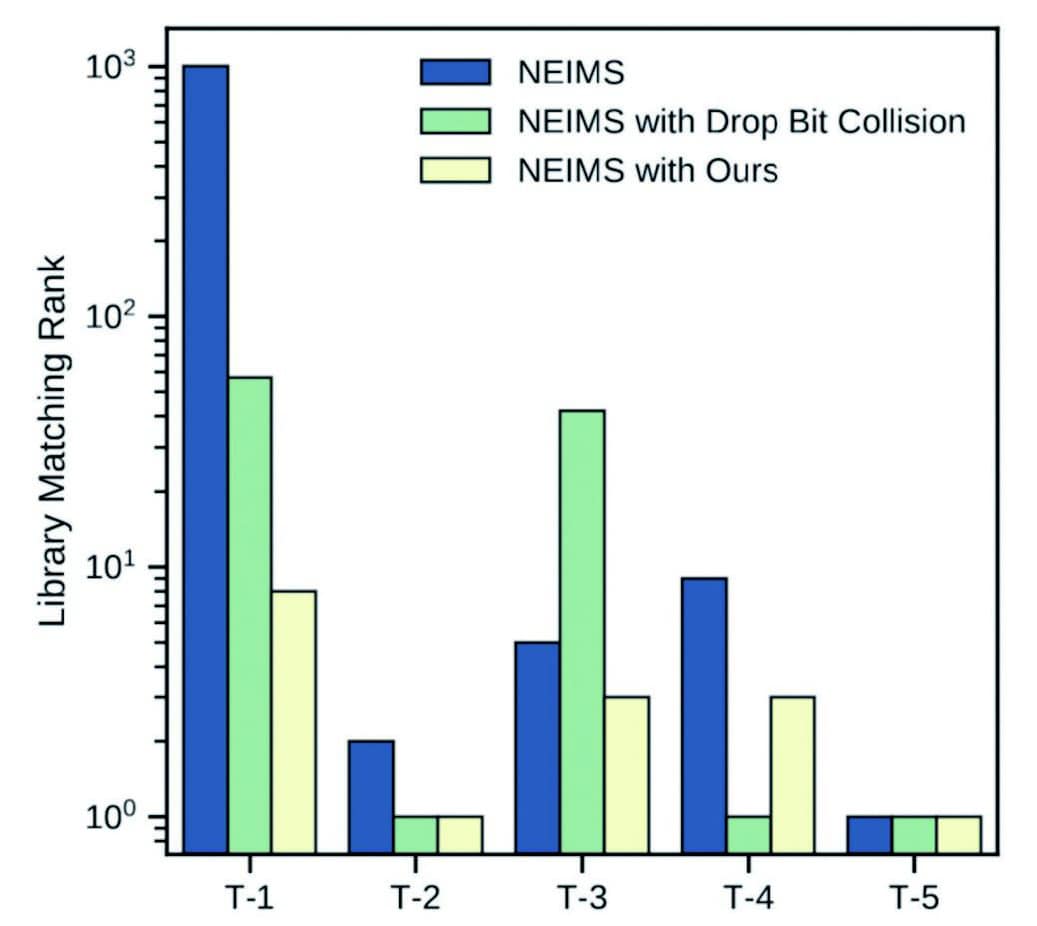 (b) Library matching rankFig.7 : Comparing the target spectra prediction scores.(a)Weighted cosine similarity.(b)Library matching rank.
(b) Library matching rankFig.7 : Comparing the target spectra prediction scores.(a)Weighted cosine similarity.(b)Library matching rank.特に全データモデルで1005位だったT-1は、997段上昇して8位であった。これは分子同定をするうえで、候補として確認することが可能な現実的な順位であり、MSSがライブラリマッチアプローチにおいて、分子同定効率を改善することを示唆している。
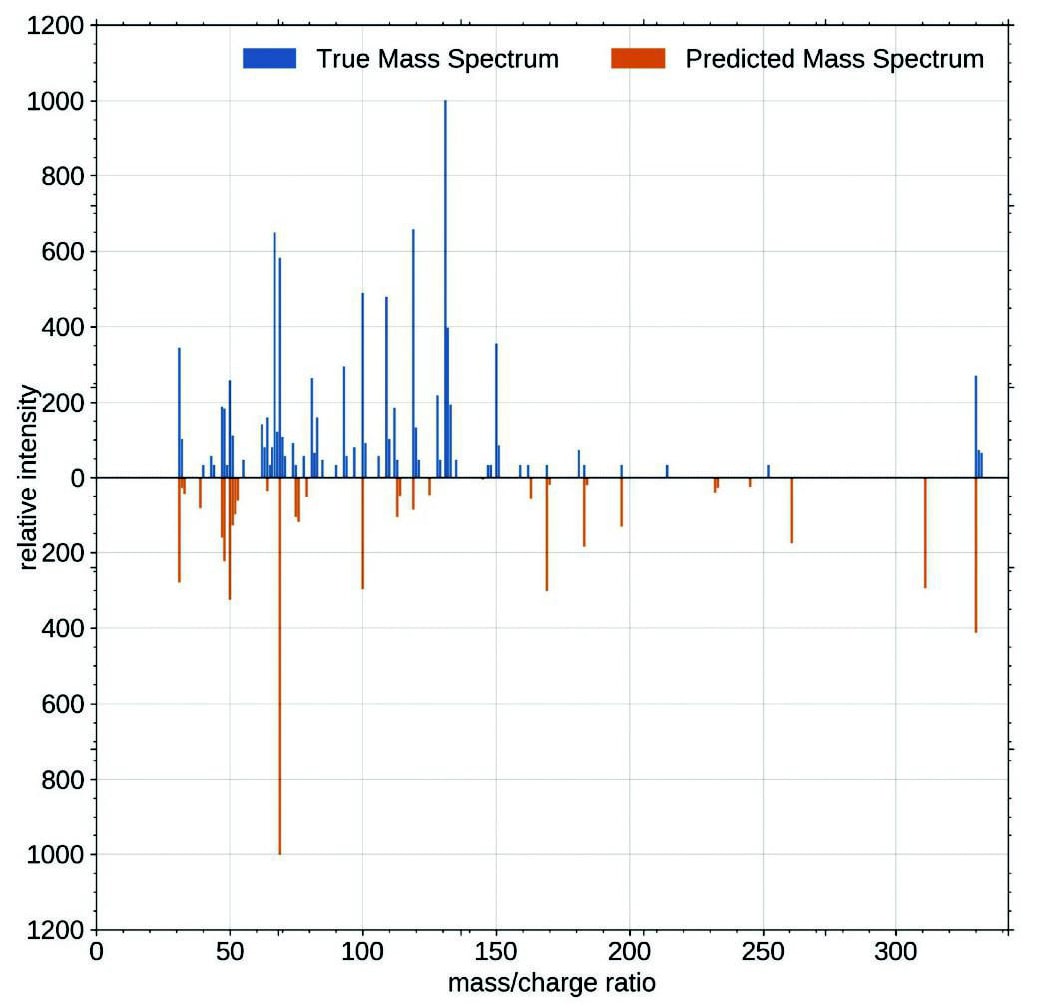 (a)
(a)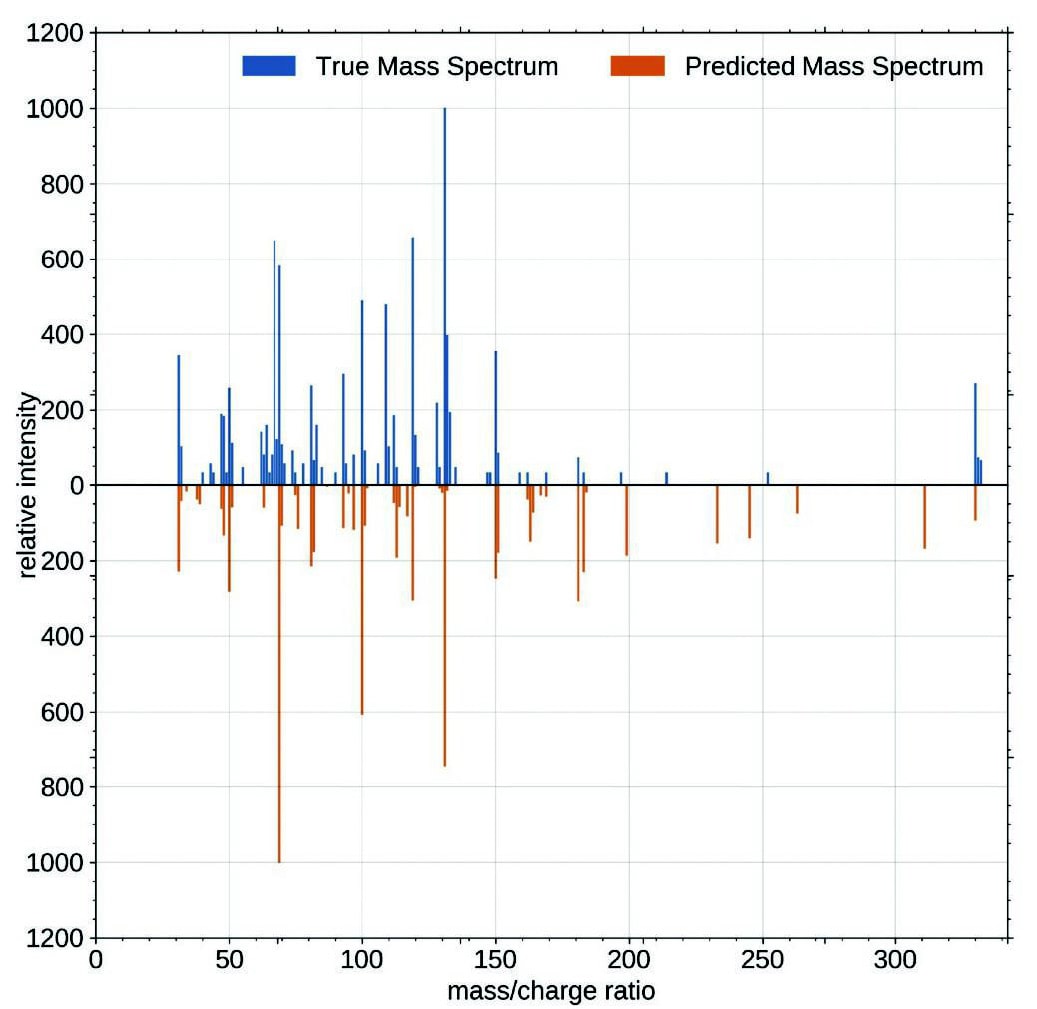 (b)Fig.8:(a)Predicted spectra with normal NEIMS, and(b)NEIMS with undersampling. The true spectrum appears in blue above, and the prediction spectrum appears in orange inverted. Normal NEIMS misses the peak at m/z = 131.
(b)Fig.8:(a)Predicted spectra with normal NEIMS, and(b)NEIMS with undersampling. The true spectrum appears in blue above, and the prediction spectrum appears in orange inverted. Normal NEIMS misses the peak at m/z = 131.T-1の予測プロット(Fig.8)を実際に見てみると、従来のNEIMSモデルはm/z=131のF2C=CF–CF2ラジカルカチオンに由来するイオンのピークが予測できていないが、我々の学習モデルは適切に予測できることが分かる。また、FF2C=CF–CF2由来イオンのピークと関係が強いと考えられるフィンガープリント(Fig.3, Identifier:644590947)のビットコリジョン発生数は57個から約25%減少していた。
ビットコリジョンがマススペクトルの予測にどう影響するかを調べるために、単純なdrop-bit-collisionモデルを検討した。drop-bit-collisionモデルは、ターゲット化合物に対してビットコリジョンが発生する化合物を除き、NISTライブラリでトレーニングされた。ビットコリジョンのみを考慮して化合物を除去したため、性能はターゲットに対して敏感であった。T-3の場合、全データモデルやアンダーサンプリングモデルと比較して類似性とランクが悪化するが、T-4では最高の類似性とランクが確認された(Fig.7)。dropbit-collisionモデルのT-3におけるコサイン類似度低下の最も大きな要因は、本来観測スペクトルには存在しないm/z=195のピークが予測スペクトルで大きく表れていることにある。また、m/z=195は、T-3から塩素を除いた構造(C5F7H2)の分子量に一致する。一般に深層学習モデルの性能劣化の原因となるデータの特定は難しく、今回のdrop-bit-collisionモデルの性能劣化の原因特定は難しい。全体的な傾向から、特定のターゲットに適したトレーニングデータから学習するように設計されたMSSアンダーサンプリングは、すべてのターゲットに対して妥当なパフォーマンスを発揮することがわかる。上記の結果は、アンダーサンプリングによってビットコリジョンがなくなるため、機械学習モデルのトレーニングが容易になることを示唆している。一部のターゲット化合物について、単純なdrop-bit-collisionよりもビットコリジョンが残っているアンダーサンプリングモデルの予測性能が優れていることは、すべてのビットを考慮して学習する全結合ニューラルネットワーク構造を持つNEIMSが、ビットコリジョンがあっても学習できることを示唆している。
学習データ全体のビットコリジョンの緩和がモデルの予測性能にどう影響するかを調べるために、単純に入力次元を増やしたモデルを用意した。その結果、次元数を増加させたモデルは既存のモデルとの性能の違いが認められなかった。この原因として、データの疎性が考えられる。入力次元を大きくすると、その結果としてデータが高次元空間に分布し、データ間の距離が離れることで疎になる。この疎なデータを用いて学習を進めると学習の進行が難しくなり、結果としてモデルの性能が向上しないと考えられる。これらの結果から、MSSがターゲット化合物の予測性能向上により効果的に対処できる可能性が示唆された。
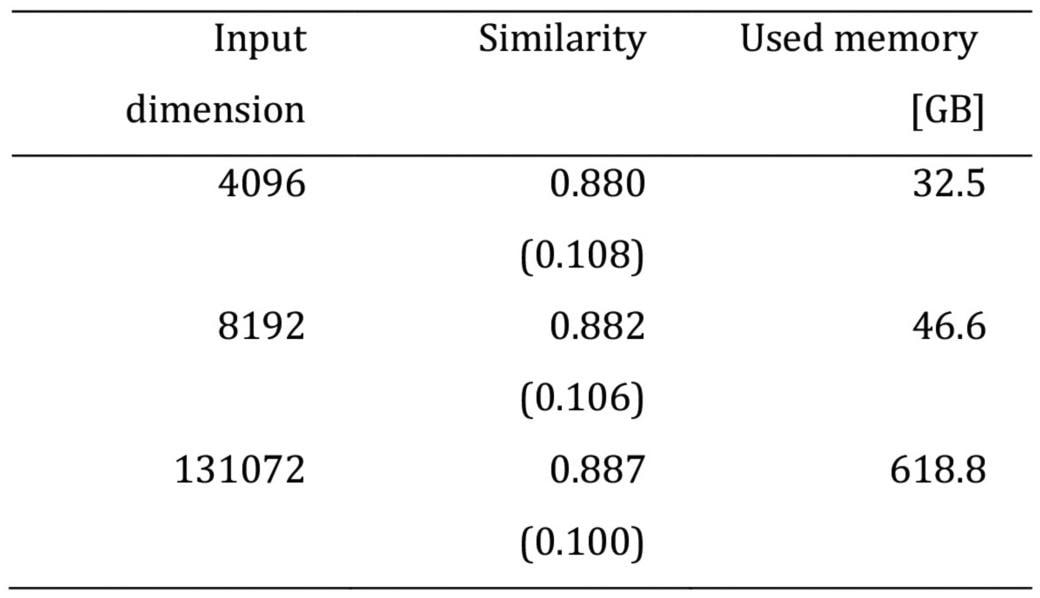
4. 総括
NEIMSベースのモデルを使用したEI-MSスペクトル予測において、マイナーな化合物に対する予測精度を向上させるために、分子類似度サンプリング:(Molecular similarity-wise sampling; MSS)を提案した。NIST ライブラリのECFPsで多数のビットコリジョンが発生しており、ビットコリジョンを軽減することでターゲットとするマイナーデータの学習が効果的に促進されることを示した。分子類似性が高いデータに対して訓練されたモデルは、ターゲット化合物と非ターゲット化合物のマススペクトルを高い精度で予測できることが実証された。本研究では、マススペクトルの予測の対象としてフッ素含有量の多い化合物を採用したが、アンダーサンプリング法は他の化合物にも適用可能であり、また、他のスペクトルや物理的性質の推定にも利用可能である。また、アンダーサンプリングは分子ビッグデータの自動クレンジング方法として期待される。単純にビットコリジョンを減らすためにベクトル長を伸ばしただけでは、データが疎となり、ResNetの学習が進まなかった。また、ビットコリジョンを解決するために入力次元を無差別に増やすと、必要なメモリ量も膨大になるため現実的ではない。グラフニューラルネットワークは、ECFPsなどの記述子を経由せずにグラフから直接学習するため、ビットコリジョンの問題もなく、改善の余地があると考えられる。実際、Zhangらは最近、グラフニューラルネットワークを使用したアーキテクチャの改善について報告した[28]。今後の研究では、MSSのグラフニューラルネットワークへの適用可能性を検討し、MSSの概念を反映した分子の類似性に基づく損失関数を設計する予定である。
5. 付録
専門用語
- アンダーサンプリング:
- 多数派クラスのデータポイントを減らすことで多数派クラスと少数派クラスのバランスを調整する。機械学習分野で使用される手法の1つで、特にクラス間の不均衡があるデータセットでよく使われる。
- オーバーサンプリング:
- 少数派クラスのデータポイントを増やすことで、少数派クラスのデータの割合を増やす手法。これにより、データセットのクラス間の不均衡を緩和し、モデルのトレーニングや予測のパフォーマンスを向上させることができる。
- ハッシュ化:
- データをハッシュ関数によって変換するプロセス。ハッシュ関数は、任意の長さのデータを固定長のバイナリ値であるハッシュ値に変換する。同じデータに対しては必ず同じハッシュ値を生成する。
- ビットコリジョン:
- ハッシュ関数において異なるデータが同じハッシュ値を生成する現象。
- radius(=2):
- 任意の原子を中心として探索する部分構造の範囲を指定するECFPsのパラメータ。radius=2ということは、中心の原子から2つの結合をたどって到達できる原子がフィンガープリントに含まれることを意味する。
- バイナリフィンガープリント:
- 分子内の特定の部分構造の存在を表すために使用される。各構造要素は0または1の値で表され、その分子に特定の部分構造が存在する場合は1、存在しない場合は0となる。
- カウントフィンガープリント:
- 分子内の特定の部分構造の出現回数を表すために使用される。各要素は整数値として表される。
参考文献
- Stein, S. E. "Mass Spectral Database". National Institute ofStandards and Technology(NIST), 2017.
- McLafferty, Fred W., and Douglas B. Stauffer. The Wiley/NBS Registry of Mass Spectral Data. Vol. 1, Wiley NewYork, 1989.
- Lai, Zijuan, et al. "Identifying Metabolites by IntegratingMetabolome Databases with Mass Spectrometry Cheminformatics". Nature Methods, vol. 15, no. 1, Nature Publishing Group, 2018, pp. 53–56.
- Grimme, Stefan. "Towards First Principles Calculation ofElectron Impact Mass Spectra of Molecules". AngewandteChemie International Edition, vol. 52, no. 24, Wiley OnlineLibrary, 2013, pp. 6306–6312.
- Bauer, Christoph Alexander, and Stefan Grimme. "How toCompute Electron Ionization Mass Spectra from FirstPrinciples". The Journal of Physical Chemistry A, vol. 120,no. 21, ACS Publications, 2016, pp. 3755–3766.
- Ásgeirsson, Vilhjálmur, et al. "Quantum Chemical Calculation of Electron Ionization Mass Spectra for GeneralOrganic and Inorganic Molecules". Chemical Science, vol.8, no. 7, Royal Society of Chemistry, 2017, pp. 4879–4895.
- Koopman, Jeroen, and Stefan Grimme. "Calculation ofElectron Ionization Mass Spectra with SemiempiricalGFNn-xTB Methods". ACS Omega, vol. 4, no. 12, ACSPublications, 2019, pp. 15120–15133.
- Koopman, Jeroen, and Stefan Grimme. "From QCEIMS toQCxMS: A tool to routinely calculate CID mass spectrausing molecular dynamics." Journal of the AmericanSociety for Mass Spectrometry 32.7, 2021, pp. 1735-1751.
- Koopman, Jeroen, and Stefan Grimme. "Calculation ofmass spectra with the QCxMS method for negatively andmultiply charged molecules." Journal of the AmericanSociety for Mass Spectrometry 33.12, 2022, pp. 2226-2242.
- Wei, Jennifer N., et al. "Rapid Prediction of Electron--Ionization Mass Spectrometry Using Neural Networks".ACS Central Science, vol. 5, no. 4, ACS Publications, 2019,pp. 700–708.
- Allen, Felicity, et al. "Computational Prediction of ElectronIonization Mass Spectra to Assist in GC/MS CompoundIdentification". Analytical Chemistry, vol. 88, no. 15, ACSPublications, 2016, pp. 7689–7697.
- Viant, Mark R., et al. "How Close Are We to CompleteAnnotation of Metabolomes?" Current Opinion in ChemicalBiology, vol. 36, Elsevier, 2017, pp. 64–69.
- He, Kaiming, et al. "Deep Residual Learning for ImageRecognition". Proceedings of the IEEE Conference onComputer Vision and Pattern Recognition, 2016, pp. 770–778.
- Rogers, David, and Mathew Hahn. "Extended-ConnectivityFingerprints". Journal of Chemical Information andModeling, vol. 50, no. 5, ACS Publications, 2010, pp. 742–754.
- Landrum, Greg, Paolo Tosco, Brian Kelley, Sriniker, et al.Rdkit/Rdkit: 2020_09_5 (Q3 2020) Release. Release_2020_09_5, Zenodo, 2021, https://doi.org10.5281/zenodo.4570805.
- Linstrom, Peter J., and William G. Mallard. "The NISTChemistry WebBook: A Chemical Data Resource on theInternet". Journal of Chemical & Engineering Data, vol. 46,no. 5, ACS Publications, 2001, pp. 1059–1063.
- Cereto-Massagué, Adrià, et al. "Molecular FingerprintSimilarity Search in Virtual Screening". Methods, vol. 71,Elsevier, 2015, pp. 58–63.
- Japkowicz, Nathalie, and Shaju Stephen. "The ClassImbalance Problem: A Systematic Study". Intelligent DataAnalysis, vol. 6, no. 5, IOS Press, 2002, pp. 429–449.
- Mazurowski, Maciej A., et al. "Training Neural NetworkClassifiers for Medical Decision Making: The Effects ofImbalanced Datasets on Classification Performance".Neural Networks, vol. 21, no. 2–3, Elsevier, 2008, pp. 427–436.
- Chawla, Nitesh V. "Data Mining for Imbalanced Datasets:An Overview". Data Mining and Knowledge DiscoveryHandbook, Springer, 2010, pp. 875–886.
- Maloof, Marcus A. "Learning When Data Sets AreImbalanced and When Costs Are Unequal and Unknown".ICML-2003 Workshop on Learning from Imbalanced DataSets II, vol. 2, 2003, pp. 2–1.
- Buda, Mateusz, et al. "A Systematic Study of the ClassImbalance Problem in Convolutional Neural Networks".Neural Networks, vol. 106, Elsevier, 2018, pp. 249–259.
- Arefeen, Md Adnan, Sumaiya Tabassum Nimi, and M.Sohel Rahman. "Neural network-based undersamplingtechniques." IEEE Transactions on Systems, Man, andCybernetics: Systems, 52.2 2020, pp. 1111-1120.
- Stein, Stephen E., and Donald R. Scott. "Optimization andTesting of Mass Spectral Library Search Algorithms forCompound Identification". Journal of the American Societyfor Mass Spectrometry, vol. 5, no. 9, Springer, 1994, pp.859–866.
- McLafferty, F. W., et al. "Probability Based Matching ofMass Spectra. Rapid Identification of Specific Compoundsin Mixtures". Organic Mass Spectrometry, vol. 9, no. 7,Wiley Online Library, 1974, pp. 690–702.
- McInnes, Leland, et al. "Umap: Uniform Manifold Approximation and Projection for Dimension Reduction". arXivPreprint arXiv : 1802. 03426, 2018.
- Jaeger, Sabrina, et al. "Mol2vec: Unsupervised MachineLearning Approach with Chemical Intuition". Journal ofChemical Information and Modeling, vol. 58, no. 1, ACSPublications, 2018, pp. 27–35.
- Zhang, Baojie, et al. "Prediction of Electron IonizationMass Spectra Based on Graph Convolutional Networks".International Journal of Mass Spectrometry, vol. 475, 2022,p. 116817.