AGC Research Report 75(2025)
Mechanism and Construction of an Exhaustive Search Method for SiO2-HF Gas Etching Catalysts Using Quantum Chemical Calculations
量子化学計算によるSiO2-HFガスエッチング触媒のメカニズム解析と網羅的探索手法の構築
飛永 諒介*
Ryosuke Tobinaga
*AGC株式会社 先端基盤研究所 共通基盤技術部 ソフトサイエンスチーム(ryosuke.tobinaga@agc.com)
本研究では、シリカガラス(a-SiO2)表面に対するフッ化水素(HF)ガスを用いたドライエッチングプロセスにおいて、ノボラック樹脂が示す触媒作用メカニズムを理論的に解明した。密度汎関数理論(DFT)計算により、ノボラック樹脂の水酸基がHF分子と強力な水素結合を形成し、HF結合を弱めることでフッ素原子の求核性を向上させ、エッチング反応の活性化障壁を低下させることを示した。この触媒作用は、従来の反応性イオンエッチング法では実現が困難であった高速かつ垂直異方性エッチングを可能にするものであり、次世代のa-SiO2デバイス製造プロセスにおける技術的革新をもたらす可能性がある。また、新規高活性触媒の探索を目的に、半経験的量子化学計算に基づく特徴量設計と線形回帰モデルを組み合わせた触媒活性予測手法を構築した。本手法の予測精度はDFT計算で見積もられた触媒活性指標と高い相関性(決定係数R2=0.89)を示し、広範囲の化合物に対して効率的なスクリーニングが可能である。高耐熱触媒を探索した事例では、62,574の分子の活性を予測し、そのうち1,410の高活性触媒候補が得られた。これらは特定の部分構造を有していることが分かり、新規触媒開発の設計指針として活用された。本研究は、a-SiO2デバイス製造におけるエッチングプロセスの効率化と高性能化を支援するだけでなく、シミュレーションと機械学習を組み合わせた触媒探索手法の有効性を示しており、幅広い分野への応用に期待される。
This study provides a theoretical explanation of the catalytic mechanism of novolac resin in the hydrofluoric acid(HF) gas-based dry etching process for silica glass(a-SiO2) surfaces. Using density functional theory(DFT) calculations, the study demonstrates that the hydroxyl groups in novolac resin form strong hydrogen bonds with HF molecules. This interaction weakens the HF bond and enhances the nucleophilicity of the fluorine atoms, ultimately lowering the activation barrier for the etching reaction. The catalytic mechanism facilitates rapid and vertical anisotropic etching, a result that has been challenging to achieve with conventional reactive ion etching methods. Consequently, it has the potential for significant advancements in the manufacturing processes of next-generation a-SiO2 devices. To identify new high-activity catalysts, a predictive method for catalytic activity was developed that combines feature design based on semi-empirical quantum chemical calculations with a linear regression model. This approach exhibits high predictive accuracy(R2 = 0.89) when compared to reference DFT calculations, enabling efficient screening across a broad range of compounds. In a case study targeting high-temperature-resistant catalysts, the active properties of 62,574 molecules were predicted, yielding 1,410 highactivity candidates. These candidates were found to possess specific substructures that provide design guidelines for developing new catalysts. This study not only enhances the efficiency and performance of etching processes in a-SiO2 device manufacturing but also underscores the effectiveness of integrating simulations and machine learning in catalyst discovery. The proposed method shows promising applications across various fields, paving the way for future advancements in catalyst development and materials science.
1. 緒言
シリカガラス(a-SiO2)は、耐薬品性、透明性、低電熱性、高周波数帯での低い誘電損失などユニークな特徴を示す1,2。近年では、a-SiO2を利用したマイクロ流体チップ3,4や、光学メタサーフェス5,6などが、次世代のデバイスとして注目されている。これらのデバイスの実用化には、a-SiO2を高速かつ垂直に微細加工するプロセスが求められる。通常、a-SiO2基板に対する異方性エッチングには、反応性イオンエッチング(Reactive Ion Etching: RIE)が用いられる7-10。しかしこれらの部材に特徴的な、深部までの垂直凹部構造や平坦な側壁はRIEによる加工では実現できていない。その主な原因は、Inductively Coupled Plasma(ICP)光源からのイオンビームがハードマスク材料をエッチングしてしまうことにある。一方で、フッ酸(HF)ガスによるSiO2表面処理は、プラズマ環境を必要としないドライエッチング法であり11-13、複数の基板の同一バッチ処理が可能である。また、微細構造間での固着の懸念が少ないため14,15、Micro-Electro-Mechanical Systems(MEMS)デバイスの製造工程の犠牲層エッチングに広く用いられている16。さらに、近年では無水HFガスに添加したアルコールがエッチング反応の触媒として機能することが報告されている17,18。
我々は、ノボラック型フォトレジストに含まれる親水性官能基が、高温環境下でHFガスとa-SiO2との反応の触媒として機能することを見出した19。この現象は、a-SiO2基板上のフォトレジストで覆われた部分が選択的にエッチングされる(Fig. 1)。これにより、従来のICP-RIEでは必須のハードマスク材料およびプラズマ源を用いることなく、垂直異方性エッチングを可能とした。また、通常のRIEと比べて約3倍の加工速度が得られるため、次世代のa-SiO2デバイスの製造における実用化プロセスとなることが期待される。
この触媒作用を示す官能基(以後、触媒基と呼ぶ)とHFガスを組み合わせた新しい垂直異方性エッチングは、触媒基を最適化することで、さらなる高速加工の実現が期待される。しかし、これまでにHFガスとa-SiO2の触媒反応機構はほとんど研究されていないため、触媒基の設計指針が明らかになっていない。
触媒設計に必要な微視的な反応機構の情報を得るには、分子シミュレーションの活用が有効である。a-SiO2はアモルファスであり、その無秩序な構造をシミュレーションすることは容易ではない。通常これらの構造は、数千~数万原子のモデルを用い分子動力学(Molecular dynamics: MD)シミュレーションなどにより求める20-22が、結合形成・解離のメカニズムを解析する計算手法では計算コストが高く非現実的なスケールである。また、局所的な反応性のみに着目し、数十原子程度のSiO2クラスターモデルを用いて密度汎関数理論(Density Functional Theory: DFT)計算によるHFとの化学反応を解析した例23があるが、実験24やMDシミュレーション25との構造の不一致が確認されることもあり、計算結果には検証が必要な場合も多い。本研究では、低い計算コストでa-SiO2の表面構造と局所的反応性の解析可能なクラスターモデルを作成した。また、そのモデルを用いてノボラック型触媒を含むa-SiO2-HFガスエッチングのDFT反応計算を実行し、触媒作用のメカニズムを検討した。
DFT計算の結果から導かれた触媒活性の支配因子をもとに、網羅的に高活性触媒を探索するためには、機械学習モデルの構築が有効である。このようなデータ駆動型の材料開発は既に様々な分野で成功を収めており、触媒探索においても盛んに研究されている26-29。今回の問題と関連が深い酸-塩基解離定数30-33や溶媒和エネルギー34-37、誘電率38, 39、有機分子の求核性40や解離エネルギー41などのパラメータは、主にランダムフォレスト42, 43やグラフニューラルネットワーク44、グラフ畳み込みニューラルネットワーク45, 46などの手法を用いて予測モデルが構築されている。しかし、触媒探索用途においては、これらのモデル構築に十分なデータが質と量の両方で不足していることが研究の大きな障壁となっており、特に実験データを必要とする例では顕著である47-50このような場合、研究者の洞察やドメイン知識、もしくはシミュレーション結果に基づく特徴量の設計が効果的である51。本研究では、DFT計算で見積もられた触媒活性予測値を参照値として、より計算負荷の低い半経験的量子化学計算に基づく特徴量設計を行った。その特徴量を用いて訓練した線形回帰モデルは高い精度(決定係数R2=0.89)でDFT参照値を予測した。また、多様な分子構造を生成する分子生成プログラムと回帰モデルを組み合わせ、触媒活性の自動予測ワークフローを構築した。
 Fig. 1 Cross-sectional SEM image of a-SiO2 glass substrate coated with photoresist-type catalyst after HF gas treatment.
Fig. 1 Cross-sectional SEM image of a-SiO2 glass substrate coated with photoresist-type catalyst after HF gas treatment.2. 計算条件
2.1. DFT計算の詳細
SiO2クラスターモデルと、HF分子および触媒基とのエッチング反応を計算するために、DFT計算を行った。全てのDFT計算は、B3LYP/6-31+G(d,p)レベル52, 53でGaussian16プログラム54を用いて実行した。触媒基としてアニオンを用いる場合のみ、その価数を電荷に指定し、その他全ての計算は電荷±0、スピン多重度1を指定し実行した。構造最適化の収束条件はGaussianプログラムのデフォルト条件55を採用した。原子ごとの電荷の値はCHELPG電荷解析56によって取得した。
遷移状態計算では、はじめに、Gaussianプログラムの遷移状態探索アルゴリズム探索57を行った。得られた遷移状態の構造に対して振動解析を行い、虚振動モードが一つであることと、その振動方向がエッチング反応の進行方向であることを確認した。次に遷移状態からIntrinsic Reaction Coordinate(IRC)計算58を実行し、虚振動の両方向に沿ったポテンシャル面の計算を行った。最後に、IRC計算の両方向の最終ステップの構造を構造最適化し、それぞれ始状態と終状態の構造として取得した。これらの構造は振動解析を行い、虚振動モードを持たない(準)安定状態であることを確認した。IRC計算の詳細については文献欄59に記載している。
2.2. 半経験的量子化学計算の詳細
回帰モデルの訓練と予測に用いる特徴量の計算のために、半経験的量子化学計算を実行した。全ての半経験的量子化学計算はxTBパッケージで実行した。全ての構造最適化計算はGFN2-xTBパラメータセット60を用いて、電荷±0、スピン多重度1を指定し実行した。最適化レベルはNormal61を採用した。原子ごとの電荷の値はMulliken解析62, 63によって取得した。
3. DFT計算を用いた触媒メカニズムの検討手法
本研究では、a-SiO2表面のHFエッチングについて、ノボラック樹脂を触媒として用いない場合(反応1)と用いる場合(反応2)を比較するために、DFT計算を用いて検討した。
計算コストを抑えるため、a-SiO2表面は水酸基で終端したβ-cristobalite(100)面を切り出したSi7O22H16クラスターモデル(以後、SiO2クラスターモデルと呼ぶ)で再現した(Fig. 2a)。SiO2クラスターモデルに対してすべての原子の自由度を考慮して構造最適化を行い、振動解析により虚振動がないことを確認した。このクラスターモデルの最適化構造は、より大きく切り出したクラスターモデルや周期境界条件をかけたスラブモデルの幾何学的構造や活性化障壁を再現していることから、局所的な反応性を説明する最小単位のモデルとして妥当であることを確認した(Table S1,S2)。また、ノボラック樹脂は、触媒基をモデル化したフェノール単分子を用いた(Fig. 2b)。
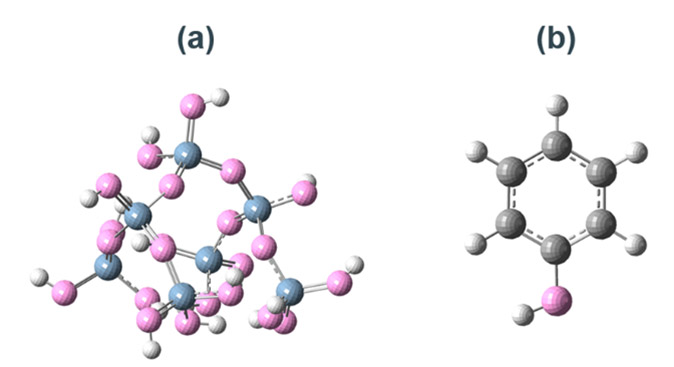 Fig. 2(a) Optimized SiO2 cluster model.(b) Optimizedphenol cluster model.
Fig. 2(a) Optimized SiO2 cluster model.(b) Optimizedphenol cluster model.4. 結果
4.1. 触媒がない場合のSiO2表面のHFエッチング
SiO2クラスターモデルからSiF4 1分子が乖離するまで、逐次的に4つのHF分子の反応プロセスを解析した結果を示す(Fig. 3a)。Fig. 4に各反応ステップにおける始状態と終状態の構造を示す。なお、Step 1、2で生成した水分子は次のステップでは系から除外した。
Step 1、Step 2の始状態で、SiO2クラスターモデルの表面にはSi-OH基とSi-O-Siの2つの異なる吸着サイトが存在する。HFの吸着エネルギーはSi-OH基の方が0.1 eV程度安定であり、優先的に吸着することが分かる(Fig. S1)。また、Si-OH基とSi-O-Siのそれぞれの結合を切断する反応の活性化障壁を比較すると、Si-OH基の方が0.1 eV程度活性化障壁が低く、反応が起こりやすい(Fig. S2)。そのため、最初の2つのHFが表面のSi-OH基をSi-F基に置換し、続く2つのHFがバルク側のSi-O-Si結合を切断する、以下の4ステップの反応が優先的に進行すると考えられる。
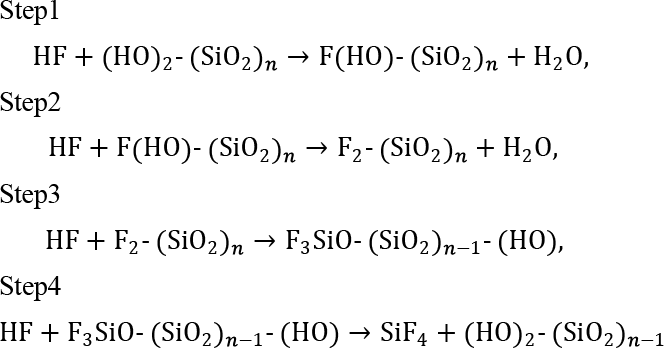
ここで、(SiO2)nはSiO2表面の構造を表し、この段階的な反応で1つのSiO2ユニットが完全にフッ素化されることを示している。各ステップの始状態では、HFはSi-OH基またはSi-O-SiのOに吸着し、安定化する。その後の遷移状態では、HFがSiにさらに近づくことでSi-O…H-Fの4員環が形成され、HFは引き伸ばされる。最終的にHとFは解離し、それぞれOへの移動とFへの求核が完了しSi-F結合が生成される。各ステップのエネルギーダイアグラムをFig. 3bに示す。Step 1が1.22 eVの活性化障壁を持つ律速反応であり、それ以降Fが配位するごとに活性化障壁は下がっていく。これはTable 1に示すように、電気陰性度の高いFがSiに配位するごとにSiの電荷密度が低くなり、新たなHFからの求核攻撃を容易に受けることが理由である。
この段階的な活性化障壁の減少とSi原子の正電荷密度の上昇は、先行の計算結果23と一致している。一方で、各ステップでの活性化障壁の絶対値が異なるのは、計算モデルや計算条件の違いであると考える。先行の計算モデルは、SiO2クラスターを水素原子で終端し、水素原子の座標を固定している。これはSi-O骨格の非晶質性を考慮しておらず、O-Si-Oの角度(177°)などでNMR実験、中性子回折実験24やMDシミュレーション25との不一致を引き起こしている(Table S1)。また、DFT計算に用いる基底関数は6-31G(d, p)を採用している。我々の事前計算では、diffuse関数を含まない基底関数が活性化エネルギーやHF分子の結合長を過大・過小評価することが明らかとなっている(Fig. S4-S6)。HF分子の結合長は、後に説明するように求核性に関わる重要なパラメータであり、先行研究は各ステップの活性化障壁を精度よく見積もれていないと考えられる。
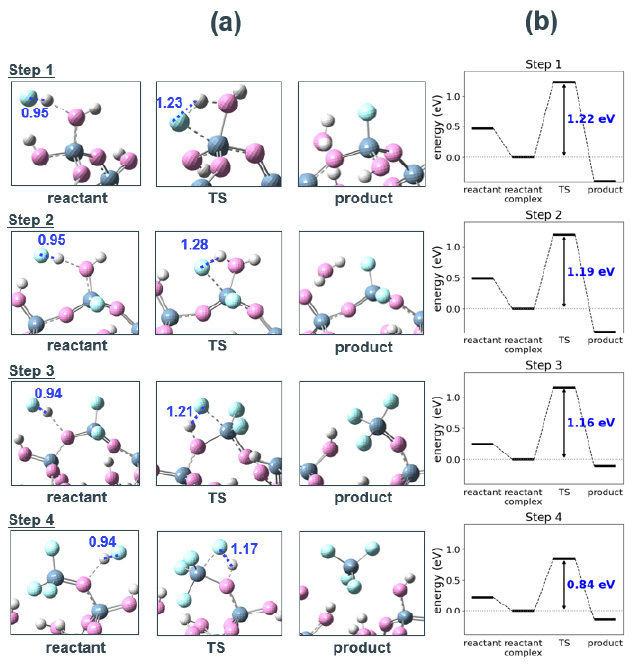 Fig. 3(a) Snapshots of the reaction process in each step for SiO2 etching by HF without phenol. The spheres are colored by atomic species.(white: hydrogen, peach: oxygen, light blue: fluorine, dark blue: silicon). Bonding distances of HF molecules are shown in blue (Å).(b) Energy diagram for each step. The blue values are the activation barriers. The vertical axis is offset by the reactant energy.
Fig. 3(a) Snapshots of the reaction process in each step for SiO2 etching by HF without phenol. The spheres are colored by atomic species.(white: hydrogen, peach: oxygen, light blue: fluorine, dark blue: silicon). Bonding distances of HF molecules are shown in blue (Å).(b) Energy diagram for each step. The blue values are the activation barriers. The vertical axis is offset by the reactant energy.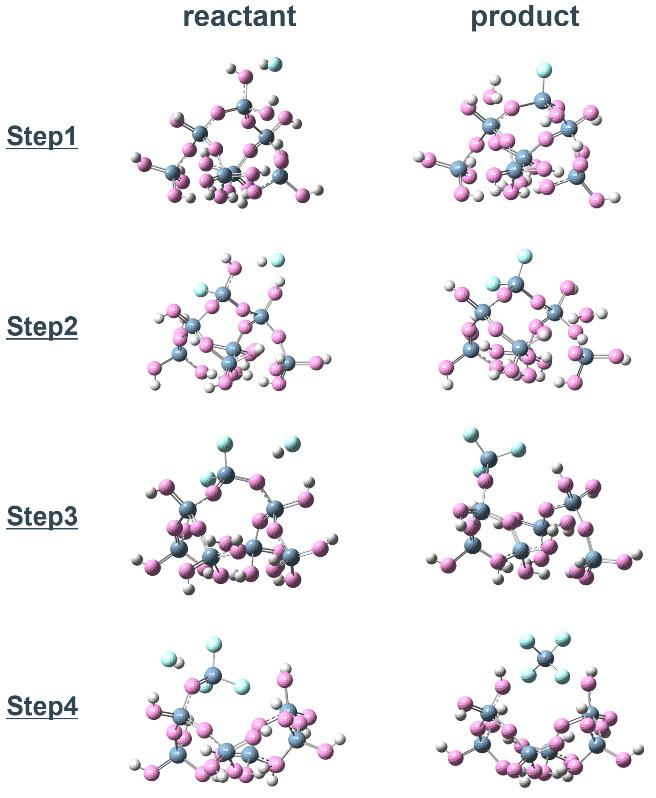 Fig. 4 Optimized structure of the reactant and product in each reaction step of SiO2 cluster -HF etching reaction.
Fig. 4 Optimized structure of the reactant and product in each reaction step of SiO2 cluster -HF etching reaction.
4.2. ノボラック樹脂を触媒に用いたSiO2表面のHFエッチング
次に、ノボラック樹脂の触媒基をモデル化したフェノール分子が存在する場合の反応を示す(Fig. 5a)。各反応ステップの始状態と終状態の構造はFig. 6に示している。各ステップの始状態は、Si-OH基(Si-OSi)に対してフェノールが吸着し、さらにフェノールの水酸基に対してHFが吸着した構造となる。これは分極の大きいフェノールの水酸基が、Si-OH基(Si-OSi)およびHFと強い水素結合を形成することで、想定される構造の中で最も安定した吸着構造となる。
Step 1の反応で比較すると、フェノールがない場合と比べて、始状態のHF結合距離は0.03 Å増加しており、HFの結合が弱まっていることが分かる。HFのFがSiへ接近すると、Si-O…H-O…H-Fの6員環が形成され、遷移状態となる。このとき、フェノールがない場合と比べてHF結合距離は0.07 Å増加している。その後、Fの求核攻撃と同時に、HFとフェノールの水酸基はプロトントランスファー機構によって、Si-Oへのプロトン移動とともにフェノールが再形成される。結果としてFig. 5bに示すように、全てのステップで活性化障壁は低減しており、律速となる障壁が1.22 eVから0.86 eVへ下がっている。これらの結果から、フェノール分子がエッチング反応の触媒として作用していることが分かり、その触媒メカニズムは、始状態から遷移状態に至るまで継続的に形成されるHFと水酸基の水素結合により、強力なHFの結合を弱める機構であることを示している。
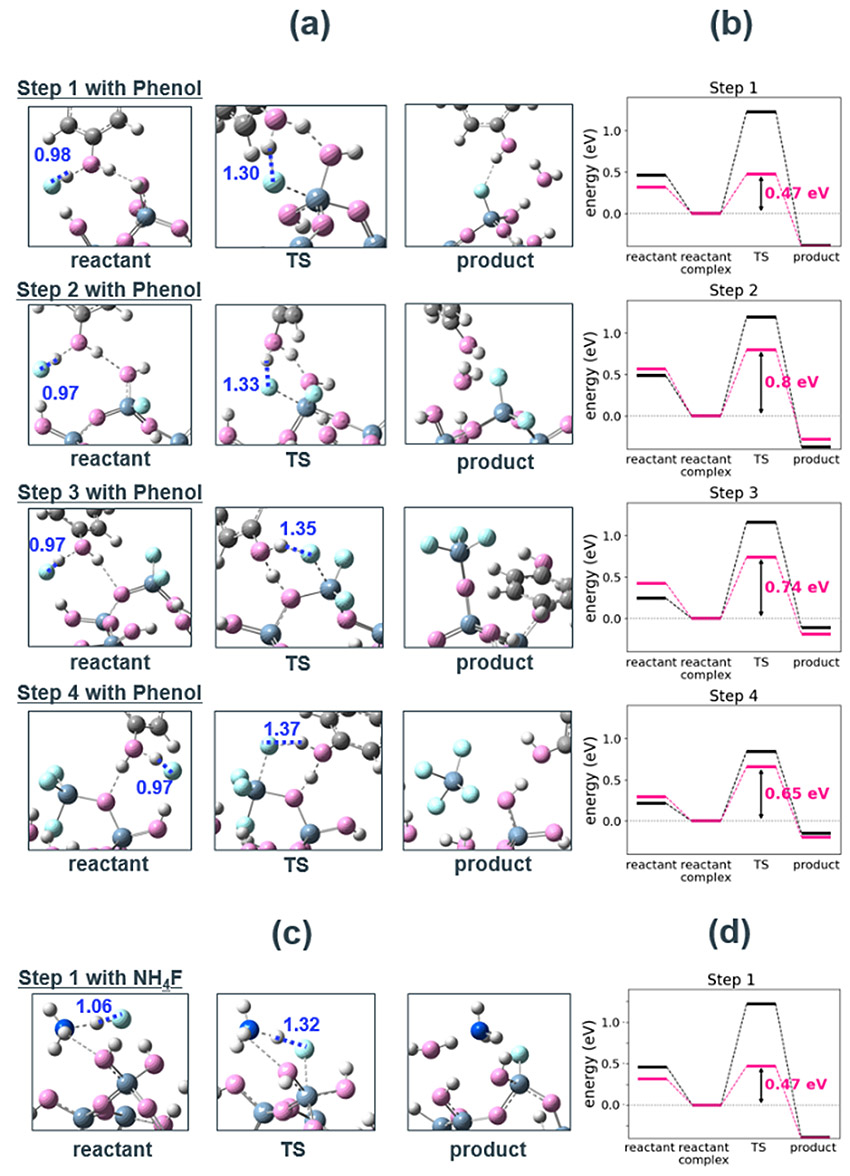 Fig. 5(a) Snapshots, and(b) energy diagrams of the reaction process in each step for SiO2 etching by HF with phenol and (c)(d) step1 with NH4F.
Fig. 5(a) Snapshots, and(b) energy diagrams of the reaction process in each step for SiO2 etching by HF with phenol and (c)(d) step1 with NH4F.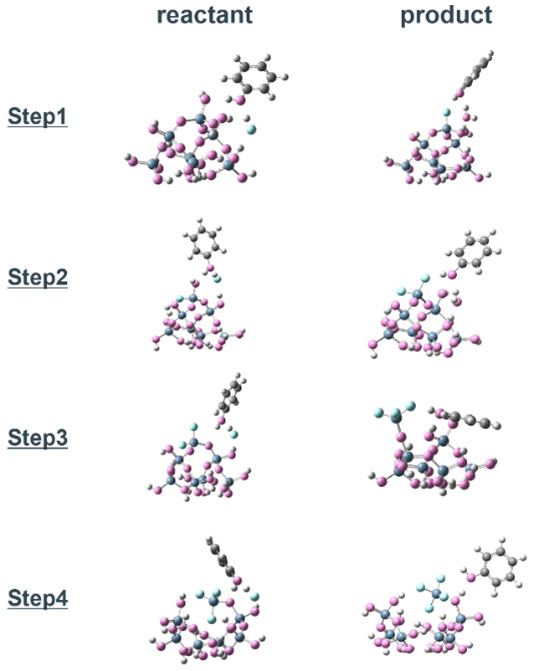 Fig. 6 Optimized structure of the reactant and product in each reaction step of SiO2 cluster -HF etching reaction with phenol catalyst.
Fig. 6 Optimized structure of the reactant and product in each reaction step of SiO2 cluster -HF etching reaction with phenol catalyst.4.3. HF結合距離による触媒効果の評価と触媒スクリーニング
4.2節の結果から、HFとの水素結合が強い触媒基は反応促進効果が大きいという仮説を立てた。その検証のために、水素結合の強さを象徴的に表す指標として、 HFと触媒基が複合体を形成した際のHF結合距離(dHF)を導入した。HFと触媒基が水素結合を形成すると、HFの水素原子は静電相互作用・ファンデルワールス相互作用により触媒基に引き寄せられる。これによりHF間の結合は弱まり、結合長は伸長する。つまり、形成される水素結合強度は、HFの結合距離の増加と相関する。フェノールよりもdHFが大きいと予測されるいくつかの分子について、DFT計算を実施し、律速段階であるStep 1の活性化障壁の大きさを比較した(Table 2)。触媒なしの場合のdHFは、構造最適化した孤立HF分子の結合長とした。触媒がない場合の活性化障壁が1.22 eV、フェノールが触媒作用した場合が0.86 eVであったのに対し、dHFが大きい分子は0.47 eV以下と、より高い触媒効果を発揮していることが分かる。フッ化アンモ二ウムNH4Fを触媒に用いたStep 1の反応過程をFig. 8に示す。フェノールと比較して、始状態と遷移状態でのHF結合距離がより増加していることから、4.2節の触媒メカニズムの考察と合致する。
計算と、それらの分子を触媒基として作用させた場合のStep 1の活性化障壁の計算を実施した。その結果、dHFの大きさと活性化障壁の大きさには負の相関が確認された(Fig. 7a)。さらに、触媒基の部分構造で触媒活性の傾向が大きく異なることが分かった(Fig. 7b)。アルコール類やエステル類と比較して、アミンやアニオンが大きく活性化障壁を下げているのは、塩基性が高く、HFとの酸-塩基相互作用が強いことが理由と考えられる。
異なるdHFを示す触媒を塗布したSiO2基板に、250℃で1000秒間のHFガス処理および処理後の重量測定を実施した実験では、dHFの大きさとエッチングレートに指数関数的な相関が確認された(Fig. 7c)。実験条件の詳細は文献19に記載されている。この結果は、活性化障壁がdHFに対して線形な相関を示していることからも理解できる。検証した触媒のうち、最も速いエッチングレートは13.39 m/minであり、従来のノボラック樹脂のエッチングレート1.00 m/minを10倍以上上回る活性を有することが分かった。
これらの結果は、dHFという1つの変数で、a-SiO2-HFガスエッチングに用いる触媒の触媒活性を評価できることを示している。加えてHF-触媒基複合体の構造最適化計算のみで評価できることは、大幅なスクリーニング効率の向上につながる。

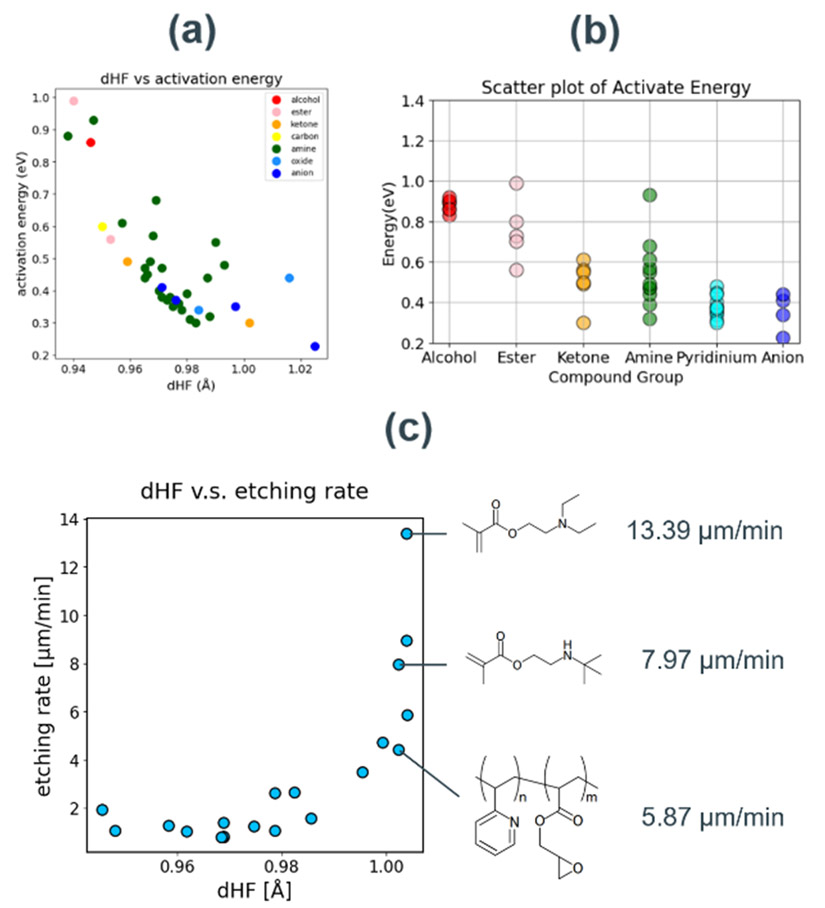 Fig. 7(a) dHF vs. activation barrier plot grouped by catalyst substructures.(b) Distribution of activation barriers for each substructure group.(c) Correlation plot between dHF and experimental etching rate. Molecular structures of highly active catalysts are explicitly illustrated.
Fig. 7(a) dHF vs. activation barrier plot grouped by catalyst substructures.(b) Distribution of activation barriers for each substructure group.(c) Correlation plot between dHF and experimental etching rate. Molecular structures of highly active catalysts are explicitly illustrated.5. 触媒活性予測手法の構築
4章では、DFT計算で見積もられたdHFが触媒活性の指標として活用できることを示した。しかし、最適な触媒基を探索するためには、多数の触媒基候補のdHFを網羅的に計算することが求められる。我々は、より高効率なスクリーニングを行うために、自動で触媒の活性予測を行うワークフローを構築した。これは、多様な分子構造を生成する分子生成プログラムと、dHFを予測する機械学習モデル(以後、dHF予測モデルと呼ぶ)から構成される。この機械学習モデルには、半経験的量子化学計算に基づく特徴量を採用し、2997分子のdHFを含むデータセットをもとに訓練した。本章では、これらの構築手順を説明する。
5.1. 分子データセットの構築
探索の中心となる触媒基は塩基性の高い窒素原子の吸着サイトを持つ化合物である。多様な塩基性分子構造を得るために、SciFinder64から10種の異なる部分構造(Table 3)を有する化合物のSMILES(SimplifiedMolecular Input Line-Entry System)65を取得した。対象の部分構造を含む化合物で、水素、炭素、窒素、酸素、フッ素、リン、硫黄、塩素、臭素、ヨウ素原子のみで構成され、総電荷が中性で、分子量350以下の単一分子を、取扱いサプライヤー数が多い順に各1000種ずつ取得した。次に、RDKit66を用いて、取得した分子の原子毎のGasteiger電荷67, 68を計算し、Gasteiger電荷が最も負に大きい原子を、HFが吸着する原子と定義し、dHFを求めた。吸着サイト原子がTable 3の10種に当てはまらない化合物は候補から除外した。また、吸着サイト原子にHF分子が吸着したとき、フッ素原子側に水素結合が形成されるような分子構造を持つ化合物を候補から除外した。酸性サイトとフッ素原子との間に形成される水素結合は大きな触媒作用を示さないため、本研究では触媒の塩基性吸着サイトによるHF分子の伸長作用のみに着目する(Fig.S7-S9)。最終的に得られた分子データの総数は2997であり、部分構造の内訳をTable 3に示す。分子データセットのd H Fを計算するためには、分子のSMILESから立体構造を生成し、吸着サイト周辺にHF分子を付加し、量子化学計算を用いて構造最適化をする必要がある。そこで、SMILESから2次元構造を生成し、明示的な水素原子を付加したのち、3次元構造を生成した。分子構造ごとに定義した吸着サイト原子に対し、余分な水素原子を一つ付加し、電荷+1eの状態で再度構造最適化を行った。得られた構造に対し、吸着サイト原子から付加した水素原子に向かうベクトルの延長線上に、フッ素原子を、水素原子から1.0 Å離して配置した。この構造を初期構造として、DFT計算による構造最適化を実施した。得られた最適化構造で、HF分子間距離が1.2 Å以上であるものや、吸着サイト原子とHF分子の水素原子との距離が2.5 Å以上であるデータを除外した。また、得られた最適化構造から見積もった候補分子とHF分子との間の相互作用エネルギーが正となった分子は、データセットから除外した。データセットの全ての構造に対して計算したdHFは、0.965 Åを中心とした正規分布状に広がっており、偏りがないことを確認した(Fig. 8)。
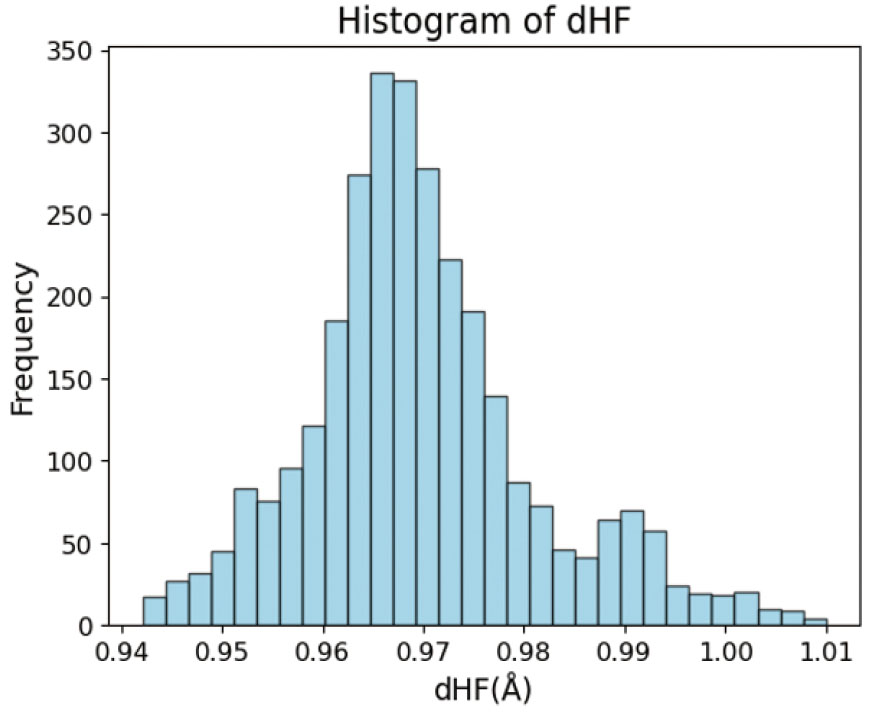 Fig. 8 Histogram of dHF of test data.
Fig. 8 Histogram of dHF of test data.5.2. dHF予測モデルの構築
5.2.1. モデルの作成条件
dHFのデータセットから生成された特徴量間の相関係数が0.8を超える場合、その他の特徴量との相関係数の総和が大きい方を除外した。これらの特徴量を追加したデータセットを、8:2の割合でトレーニングデータとテストデータに分割した。さらに、RパッケージBoruta(v8.0.0)のBoruta関数69を使用して、特徴選択を実行した。特徴量選択におけるランダムシードの選択は、予測精度に大きな影響を与えなかったため、シード0を使用し、最大iteration数を200として実行した。Iterationが最大に達した時点で暫定状態の特徴量は除外した。また、これらの特徴量を、平均値が0、分散が1になるように標準化した。この手順で選択した特徴量のみをラベル付けしたトレーニングデータを用いて、Scikit-learn70で実装したランダムフォレスト回帰モデル、線形回帰モデル、Ridge回帰モデル71、Lasso回帰モデル72を訓練した。ランダムフォレストモデルの決定木の数やランダムシードの選択は予測精度に大きな影響を与えなかったため、それぞれ100、および0と設定した。Ridge回帰モデルとLasso回帰モデルにおける損失関数のパラメータα値は、グリッドサーチにより決定係数(R2)が最も高くなる値を使用した。各モデルのその他のハイパーパラメータはデフォルト値を採用した。
5.2.2. 特徴量設計とモデルの性能評価
はじめに、汎用的な特徴量セットであるRDKit記述子とMordred記述子73を用いて精度検証を行った。特徴量の総数は45と17であった。評価指標と参照値-予測値プロットをTable 4、 Fig. 9に示す。最も高いR2は、Mordred記述子を用いたランダムフォレストモデルが示した0.70であり予測精度は低い。これは、これらの特徴量セットが分子構造全体の性質を表すことに起因する。例えば重要度が最も高い特徴量はそれぞれBCUT2D_MRHIとnBaseであり、これらは、分子内の原子の最も高い分極率を表す特徴量と、塩基性部分構造の数をカウントする特徴量である。4.3節で説明した通り、dHFは吸着サイト部分の塩基性が重要であるため、局所構造に特化した特徴量を設計する必要がある。
分子の局所的な性質を表す簡易な指標は、構成原子の部分電荷である。そこで、Gasteiger法を用いて計算したところ、部分構造の種類毎にシフトしているものの、吸着サイト原子の部分電荷はdHFと精度よく相関している(Fig. 10a)。この部分構造種ごとの異なる傾向を補正するために、吸着サイト原子の原子種や、吸着サイト原子近傍の構造、吸着サイト原子近傍のGasteiger電荷などを加えた特徴量セット(以後、Gasteiger特徴量と呼ぶ)を作成した。前節で説明した方法で、特徴量の総数を37個に削減した。Gasteiger特徴量で訓練したランダムフォレストモデルの評価指標と参照値-予測値プロットをTable 4、Fig. 10bに示す。汎用記述子に比べ、R2は0.88と大きく向上しているものの、アミン以外のデータ(O、P、S)は予測精度が低いことが分かる。これは、アミンに対しては詳細な構造分類を設定しているものの、それ以外の構造は原子種のみ考慮していることが理由である。つまりこのモデルは、探索範囲の全ての構造に対して事前に特徴量を設計する必要があり、想定外の構造に対しては予測が不可能である。一方で、アミンだけを予測対象にした場合のR2は0.91と非常に高く、部分構造による予測精度の差も少ないことから、探索範囲が定まっている問題に対しては有効なモデルであると言える。
Gasteiger特徴量を用いた予測結果の向上に構造分類が必須であるのは、Gasteiger法が原子種の電気陰性度のみで電荷を計算することに由来する。あらゆる部分構造に対しても汎用に予測をするためには、高次の電荷計算手法や構造最適化による結合長、エネルギーの情報が必要である。一方でDFTや波動関数理論に代表される量子化学計算手法は、探索における大幅な律速となることが懸念される。これらの課題を解決する方法として、半経験的量子化学計算を活用した特徴量設計と、それに基づく予測モデルの性能検証を行った。
汎用な半経験的量子化学計算パッケージであるxTB74, 75は、DFT計算のボトルネックを半経験的ポテンシャル76, 77に置き換えることで、高速かつ妥当な精度の計算手法を実装している。以前のベンチマークでは、分極DZ基底レベルのDFT計算と同様の1電子特性を提供しつつ、3~4桁程度高速であることが報告されている78。この精度と速度のバランスにより、物性予測のための機械学習モデルの特徴量として用いられており、例えば有機分子の光学応答79や酸-塩基解離定数80、モノマーの動的性質81の予測に活用されている。
特徴量の設計には、xTBを用いて最適化した構造の電荷分布、結合長、エネルギーの値を用いた。はじめに、xTBを用いた構造最適化でdHFを計算し、DFT計算を用いた参照dHFとの比較を行った。xTBで計算したdHFは、DFT計算と比較して0.005 Å程度過大評価するが、0.98 Å以下の短いdHF領域では線形に相関していることが分かる(Fig. 11a)。一方で長いdHF領域では最大0.02 Å過大評価し、傾向が異なることが分かった。この値をHFs_xTBという特徴量に定義し、単回帰分析を実行したところ、R2は0.71であった。そこで、全体的なばらつきや、特に長いdHF領域の異なる傾向を補正するため、追加の特徴量設計を行った。
xTBで構造最適化した触媒基-HF分子複合体の、HF分子の水素原子と吸着サイト原子の距離を表すdistances_HB_xTBを作成した。この距離は、dHFと同様に水素結合の強度に相当するが、長いdHF領域で目的変数との相関が高い。次に、エネルギーの観点から水素結合強度を説明する値として、触媒基とHF分子の相互作用エネルギーを表すads_xTBと、触媒基のプロトン親和力を表すPAs_xTBを作成した。これらは、目的変数との直接的な相関は弱いものの、立体反発の影響や吸着サイト原子以外からの弱い相互作用を説明する可能性がある。次に、電荷の情報として、charges_site_ads_xTB、charges_site_d_xTB、charges_F_xTB、charges_proton_xTBを追加した。これらはそれぞれ、触媒基-HF分子複合体の吸着サイト原子の電荷、触媒基-HF分子複合体と孤立触媒基の吸着サイト原子の差電荷、触媒基-HF分子複合体のHF分子のフッ素原子の電荷、触媒基の吸着サイト原子が水素化した共役酸の水素原子の電荷を表す。このような電荷の情報は、吸着サイト原子から相互作用する酸への電荷移動量、つまり電子供与性を表し、電子移動の観点からdHFを説明する特徴量になりうると考えた。また、4つのGasteiger特徴量をxTBで再計算し、採用した。以上の物理的に意味が明確な12の特徴量を用いて回帰モデルを構築した。
xTB特徴量セットを用いて訓練したそれぞれのモデルの評価指標と参照値-予測値プロットを示す(Table 4、 Fig. 11b, c)。最も高いR2は、ランダムフォレストモデルの0.94であり、Gasteiger特徴量に比べて向上した。最も重要度の高い特徴量はHFs_xTであり、半経験的手法を用いた構造最適化がDFT計算の結果を良く再現していることが大きな理由である。また、Fig. 11aと比較すると、全体的なばらつきや長いdHF領域の予測精度が改善しており、原子種や部分構造による予測精度の偏りもない。これは、結合長や電荷、エネルギーの情報を加えて包括的な特徴量セットを構築していることが理由である。線形回帰モデルのR2がランダムフォレストに劣るのは、一部の特徴量が目的変数に対して非線形に相関することが理由であるが、R2は0.89であり、部分構造による精度差もないことから十分な精度である。Ridge回帰やLasso回帰による精度向上は見られなかった。
次に、テストデータに含まれない部分構造の予測精度を検証した。窒素原子を吸着サイト原子に持つ構造(aroma6~1-amine)だけを8:2の割合で訓練データとテストデータに分割した。さらに、その他の全ての構造(O、P、S)をテストデータのみに加え、モデルの訓練と予測を行った。結果、外挿探索で悪い結果を与えやすい82ランダムフォレストモデルのR2は0.81で、大きく低減したのに対し、線形回帰モデルのR2は0.89で変化がなかった。Fig. 11dに示す参照値-予測値プロットは、アミン類のデータのみで訓練したランダムフォレストモデルがその他の部分構造(O、P、S)を説明できていないことを表している。線形回帰モデルは、訓練データに含まれていない構造に対して最大0.02 Åの誤差を生じているが、大半の構造のdHFを予測できている(Fig. 11e)。この予測精度は、膨大な候補が存在する有機触媒探索において、最初のスクリーニングを行うのに十分である。この特徴量セットに含まれる全ての特徴量は、xTBで最適化された触媒基や触媒基-HF分子複合体の構造、電荷、相対エネルギーから導かれる値であり、分子構造に依存する補正や分類を必要としない。そのため、特徴量の計算や後処理に人為的な操作を介さず、次章に説明する分子生成プログラムと組み合わせることで、触媒活性自動予測ワークフローを構築することが可能である。

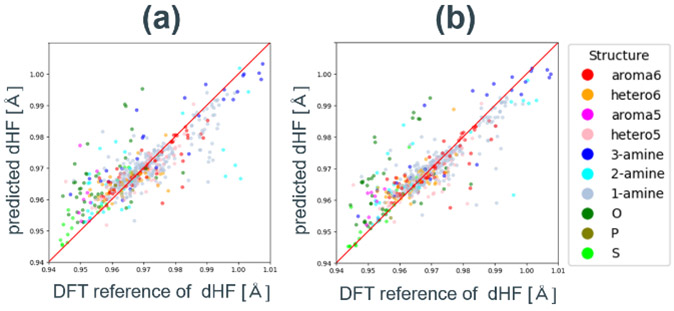 Fig. 9 DFT reference vs. predicted dHF plots for random forest models with different feature sets.(a) RDKit features reduced by Boruta.(b) Mordred features reduced by Boruta.
Fig. 9 DFT reference vs. predicted dHF plots for random forest models with different feature sets.(a) RDKit features reduced by Boruta.(b) Mordred features reduced by Boruta.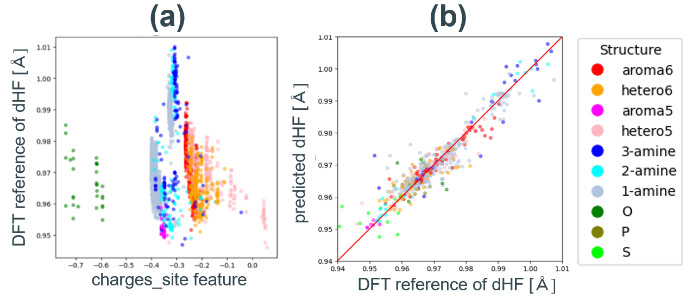 Fig. 10(a) Charge_site feature vs. DFT reference plot.(b) DFT reference vs. predicted dHF plots for random forest models with Gasteiger charge based feature set.
Fig. 10(a) Charge_site feature vs. DFT reference plot.(b) DFT reference vs. predicted dHF plots for random forest models with Gasteiger charge based feature set.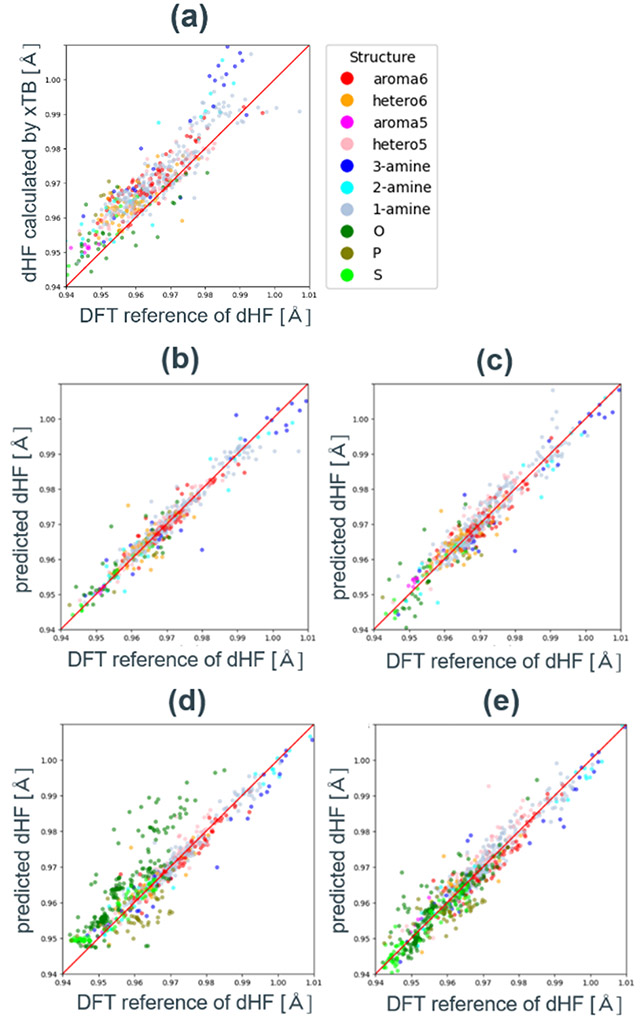 Fig. 11( a) dHF correlation plot calculated by DFT and xTB. DFT reference vs. predicted dHF plots with xTB based feature set with (b) random forest model,(c) linear regression model,(d) random forest model trained with only amine data and (e) liner regression model trained with only amine data.
Fig. 11( a) dHF correlation plot calculated by DFT and xTB. DFT reference vs. predicted dHF plots with xTB based feature set with (b) random forest model,(c) linear regression model,(d) random forest model trained with only amine data and (e) liner regression model trained with only amine data.5.3. 分子生成プログラムの実装
dHF予測モデルに読み込ませるSMILES入力データを自動で生成するプログラムを実装した。分子生成アルゴリズムとして広く知られているのは遺伝的アルゴリズム83-85やグラフニューラルネットワークであり、これまで、特定の励起波長を示す材料設計86や、特定の太陽電池ドナー分子と適合するアクセプター分子探索87、分割構造の学習による合理的な分子設計88などが提案されてきた。これらの手法は、構造と目的変数の関係が明らかでない場合のよい解決策であり、また、想定外の特殊な構造を取得できる利点もある。
本研究の場合、探索の対象は比較的明確であり、窒素、酸素、リン、硫黄などの塩基性吸着サイトを含み、その近傍に多様な部分構造を有する有機分子である。また、得られた触媒候補は将来的に産業応用を目的として検証されるため、特殊な構造を有する材料よりも汎用的な材料が望ましい。これらの理由から、汎用的な初期構造をベースに部分構造を改変するルールベースの分子生成プログラムを実装した。実装したプログラムの分子生成フローをFig. 12に示す。
はじめに、初期構造のSMILESを読み込む。この初期構造に、RDKitのAddHsメソッドを用いて明示的な水素原子を付加する。次に、初期構造に含まれるダミー原子もしくはランダムな一つの水素原子を対象に、以下のいずれかの構造の改変を確率的に実行する。
( i )1原子の追加
(ⅱ)二重結合の追加
(ⅲ)6員芳香環の追加
(ⅳ)5員芳香環の追加
(ⅴ)脂肪族環の追加
ここで追加する原子や環は、指定された原子種に確率的に置換される。また、この改変を一度行う度に、分子構造に明示的な水素原子を付加し、
①分子構造の形式電荷が0であること
②ケクレ構造であること
③3次元構造が生成可能であること
を確認した。これらを満たした分子構造は繰り返し構造の改変が行われ、指定の分子量を超えた場合、その直前の改変によって生成された分子構造が保存される。構造の改変の途中で誤った分子構造と判定された場合、その分子構造を削除し、再度初期構造から構造改変を開始する。
このようにして実装したプログラムはFig. 13に示すように、初期構造を含む多様な分子構造を生成可能であることを確認した。また、特徴量計算や予測の律速にならない十分な速度で分子構造を生成可能であることを確認した。
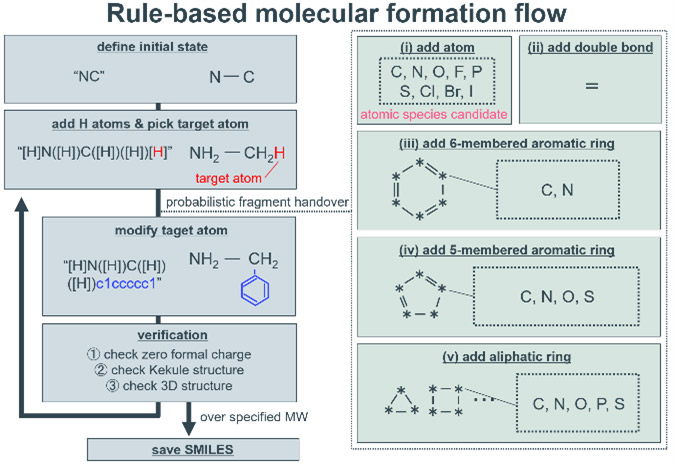 Fig. 12 Schematic of rule-based molecule formation program.
Fig. 12 Schematic of rule-based molecule formation program.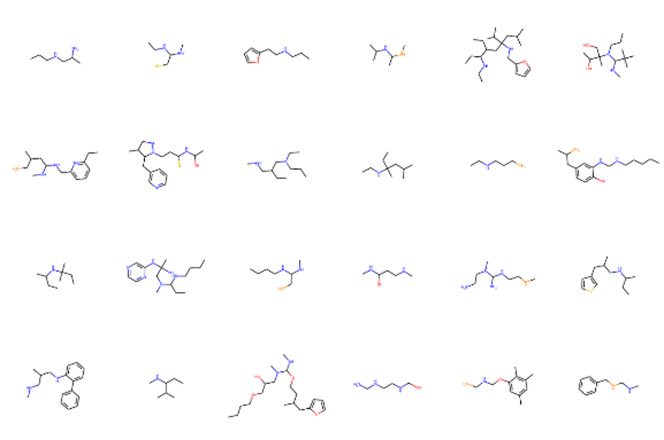 Fig. 13 Example of molecular structure generated with methylamine as the initial structure.
Fig. 13 Example of molecular structure generated with methylamine as the initial structure.5.4. ワークフローの実装
実装した触媒活性予測ワークフローの概略図をFig. 14に示す。
はじめに、分子生成プログラムによって初期構造を含む分子のSMILESを生成する。そのSMILESを読み込み、xTBを用いて特徴量計算に必要な構造最適化と電荷解析を実行する。それぞれの最適化構造が指定の範囲内の結合長やエネルギーを有する場合のみ特徴量計算を行う(Table S3)。特徴量を読み込み、事前に訓練したdHF予測モデルでdHFの予測を実行する。予測したdHFが1.00 Å以上であった場合、DFT計算で再度構造最適化を実行する。DFT計算が実行された分子は、データが保存され、dHF予測モデルの訓練データに加えられる。この追加の訓練は、触媒探索に特に重要な高活性領域や外挿領域の将来的な予測精度向上を図ったものであり、現時点では十分なデータ追加ができていないため、本稿では説明を省略する。このようにして、多様な触媒分子の性能を自動で評価しつつ、モデルの訓練データを逐次追加するワークフローを構築した。
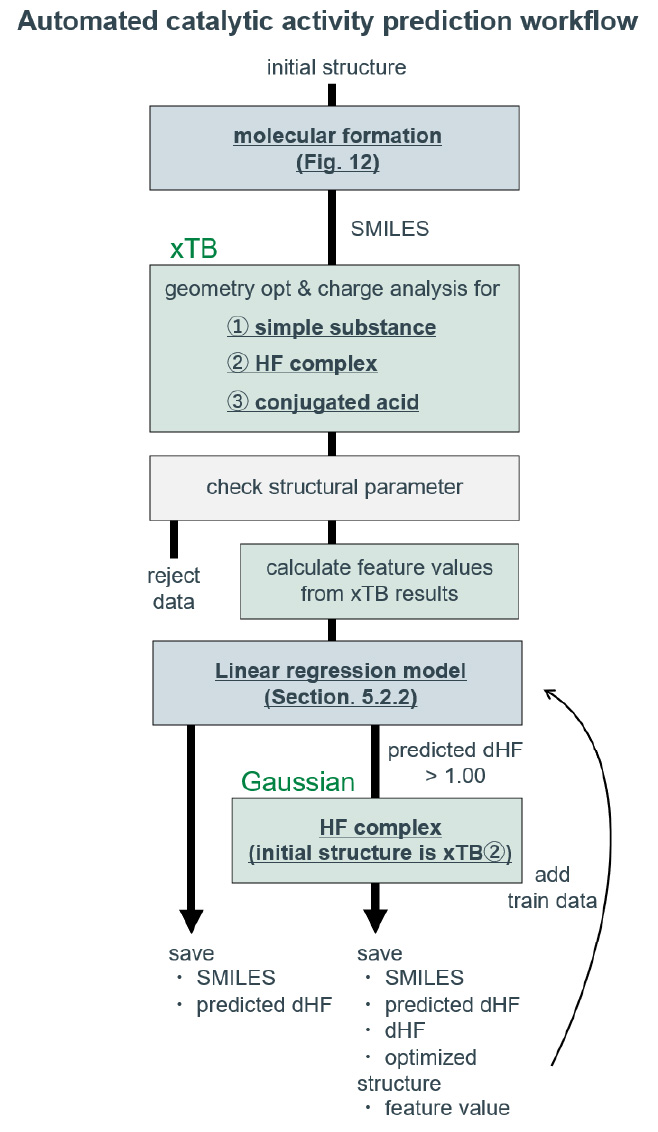 Fig. 14 Schematic diagram of the catalytic activity prediction workflow.
Fig. 14 Schematic diagram of the catalytic activity prediction workflow.6. 触媒探索スキームの活用例:高活性耐熱触媒の探索
これまで触媒基が有する触媒活性を議論してきたが、実用的な触媒を開発するには、ノボラック樹脂のように触媒基を含むポリマーを考える必要がある。実験からポリマー特性がエッチング形状に影響を与えることが分かっている。例えば、ともに水酸基を有するノボラック樹脂とPolyglycerol Polyglycidyl Ether(PPE)はそれぞれdHFが0.956と0.960であり、同等の触媒活性を有する。一方で、これらをパターニングしHFガス処理をした実験89では、加工形状が大きく異なることが分かっている。ノボラック樹脂を用いた加工は底部がやや湾曲しているものの、小さいテーパー角と平坦な側壁を有する(Fig. 15a, b)。PPEを用いた加工は、エッチングの方向が一定でなく、大きく変形していることが分かる(Fig. 15b, c)。この加工形状の違いは、触媒分子の熱分解温度に起因する。高温HFガスで処理するという本プロセスの特性上、熱分解温度が低いPPEは、エッチングの進行と同時に触媒の分解と変形、それに伴う反応の不均一化が起こっている。これらの結果から、触媒活性が高く、かつ耐熱性も高い触媒分子の開発が急務となっている。
耐熱性の高いポリマー材料として代表的なのが、ポリベンゾオキサゾール(Polybenzoxazole: PBO)類である(Fig. 16a, b)。PBOは、良好な機械特性、優れた熱安定性および優れた耐環境性について、過去数十年研究されており90-92、その熱分解温度は350 ℃~650 ℃程度である93, 94。これは、SiO2-HFガスエッチングのフッ化水素雰囲気温度250 ℃を優に超える値であり、耐熱触媒としての応用が期待される。一方で、その触媒基であるベンゾオキサゾール(Benzoxazole:BO)のdHFは0.970であり、触媒活性はそれほど高くはない。そのため、BOの分子骨格であり、かつ活性が高い触媒基を探索する必要がある。この課題を解決するために、BO骨格(Fig. 16c)を初期構造として、触媒活性予測ワークフローを実行した。
回帰モデルに読み込まれた全ての分子の予測dHFの分布をFig. 17に示す。BO骨格に対して、部分構造を改変することで、より高いdHFを示す触媒分子が多数得られた。予測した分子数は重複を除き62,574であり、そのうちdHFが0.985 Å以上の高活性触媒候補は1,410であった。本モデルは合成難易度を考慮していないため、高いdHF予測値を示した候補から合成可能な分子を抽出する必要がある。今回はより効率的に合成候補を決定するため、得られた分布の3段階のピークをもとに、高い活性の要因を調査した。この3段階の異なる触媒活性は、部分構造の特徴に基づくと仮説を立て、Uniform Manifold Approximation andProjection(UMAP)95を用いて2次元マップによる可視化を試みた。今回の予測の2次元マップでは、類似分子群の集合領域と予測活性の程度が明確に対応付けられることが分かった(Fig. 18)。予測活性が高い領域はBOの両端が長い炭素鎖で構成されている分子群であり、予測活性が中程度の領域は炭素鎖の途中に芳香環やヘテロ原子を含む分子群である。予測活性が低い領域は芳香環やヘテロ原子が直接結合している分子群であることが分かった。要するに、これらの類縁体の触媒活性は、BO環内に電子を供与するか、吸引するかで説明することができ、電子供与性が高いアルキル鎖を有する構造が最も高活性であると結論づけた。
これらの結果から、PBO類のポリマーに、アルキル鎖のスペーサーや側鎖を導入することで、高活性を付与することが可能であると考える(Fig. 19)。しかしながら、これらのポリマーがPBOと同等の耐熱性を有しているかは検証が必要である。PBO類は未だパターニング手法が確立しておらず、性能評価が可能なプロセス技術の開発が今後の重要な課題となっている。
 Fig. 15 SEM images after HF gas treatment using (a)(b) novolac resin and (c)(d) PPE as catalyst89.
Fig. 15 SEM images after HF gas treatment using (a)(b) novolac resin and (c)(d) PPE as catalyst89.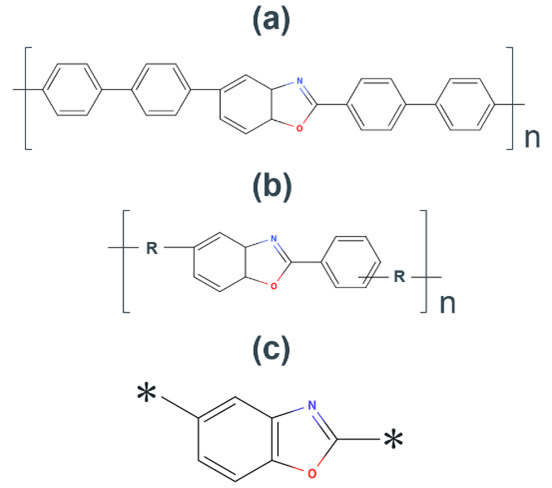 Fig. 16 Molecular structure of(a) PBO,(b) PBO polymerized by π-stack and(c) BO skeleton.
Fig. 16 Molecular structure of(a) PBO,(b) PBO polymerized by π-stack and(c) BO skeleton.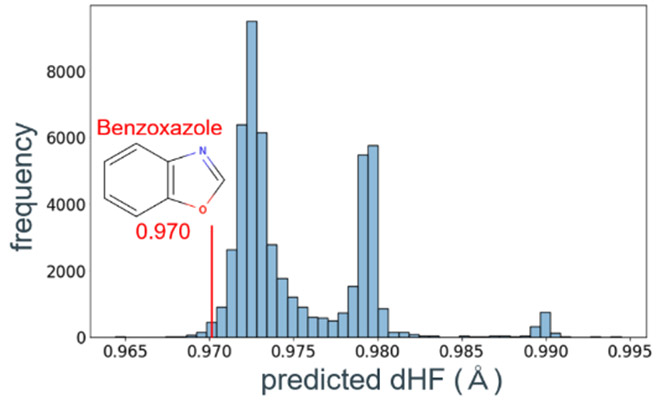 Fig. 17 Histogram of predicted dHF of molecular structures with benzoxazole skeleton.
Fig. 17 Histogram of predicted dHF of molecular structures with benzoxazole skeleton.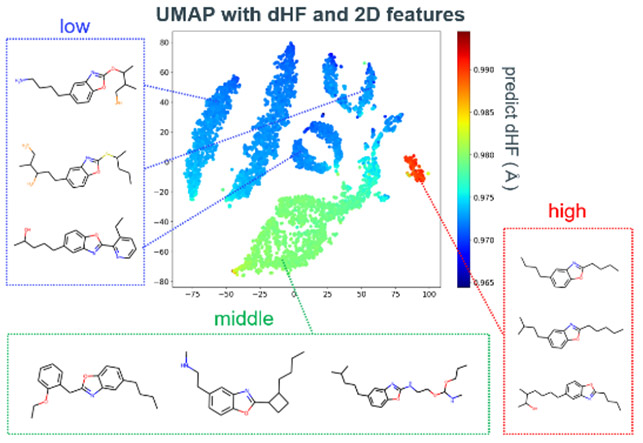 Fig. 18 UMAP based on predicted dHF and 2D features and representative structures of each region.
Fig. 18 UMAP based on predicted dHF and 2D features and representative structures of each region.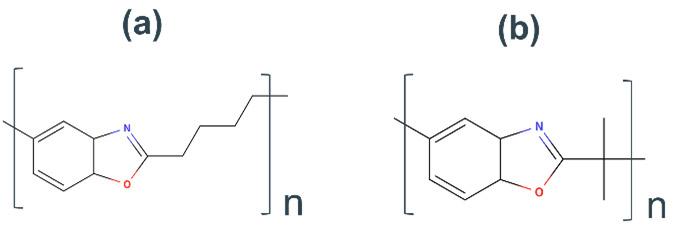 Fig. 19 Example structure of benzoxazole polymers predicted to be highly catalytic activity. Polybenzoxazole with(a) Carbon chain spacers and(b) side chains imparted.
Fig. 19 Example structure of benzoxazole polymers predicted to be highly catalytic activity. Polybenzoxazole with(a) Carbon chain spacers and(b) side chains imparted.7. まとめ
本研究では、DFT計算を用いて、a-SiO2表面のHFドライエッチングにおけるノボラック樹脂の反応促進機構を解明した。ノボラック樹脂の水酸基のように分極した触媒基は、HF分子と強い水素結合を形成し、強力なHF結合を弱める効果がある。その結果、求核性が向上したフッ素原子がケイ素原子へ容易に攻撃ができ、エッチング反応の活性化障壁が下がることが分かった。この触媒基の活性の高さは水素結合の強度と相関するため、触媒基と相互作用したHF分子の結合長を指標にして効率的に高活性触媒をスクリーニング可能であることを示した。
また、高活性触媒を効率よく、かつ網羅的に探索するために、分子自動生成プログラムと線形回帰モデルを組み合わせた触媒活性予測ワークフローを構築した。回帰モデルの構築では、様々な回帰モデルの手法や特徴量を比較し、最も高い予測精度を示した半経験的量子化学計算に基づく特徴量を採用した。このモデルはDFT計算により導かれる触媒活性指標をR2=0.89の精度で予測可能であり、データセットに含まれていない化合物も探索できる。
本稿では、高活性耐熱性触媒の探索において本スキームを活用した例を紹介した。高耐熱分子骨格を有するという制約のもと、62,574の分子構造の触媒活性を予測し、1,410の高活性触媒候補が得られた。さらに、有用な分子骨格による付加性能を考慮することや、特徴量の追加によって合成難易度や価格によるスクリーニングにも応用が可能である。これらの結果は、次世代のa-SiO2デバイス製造における垂直異方性エッチングプロセスの大幅な効率化、高性能化を実現する可能性を示している。
本研究で提案した触媒活性予測ワークフローは、高効率かつ網羅的な触媒探索を可能とし、実験検証や訓練データの追加を通じてさらに精度を向上可能なスキームであり、無機材料や有機金属錯体への展開が期待できる。本研究で得られた知見は、シミュレーションと機械学習を組み合わせた材料開発の有効性を示しており、a-SiO2エッチング技術の革新にとどまらず、幅広い分野での応用が期待される。
付録
Si-O-Siへのエッチング反応
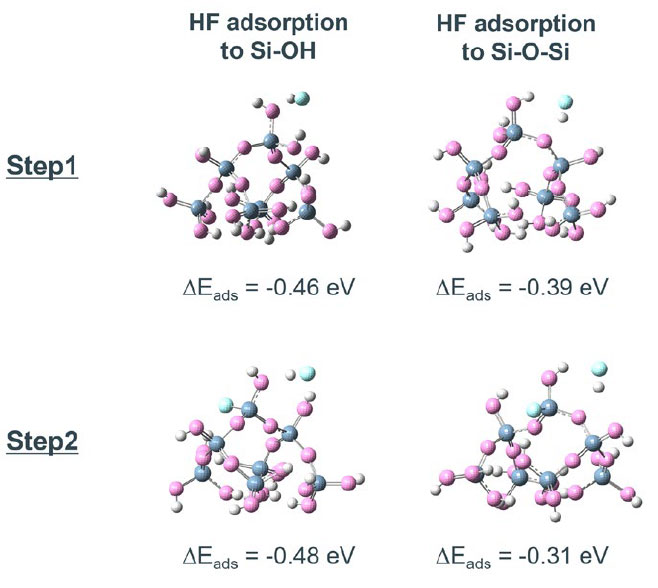 Fig. S1 Adsorption structures of HF to different O site on SiO2 cluster model. ΔEads represent the respective adsorption energies.
Fig. S1 Adsorption structures of HF to different O site on SiO2 cluster model. ΔEads represent the respective adsorption energies.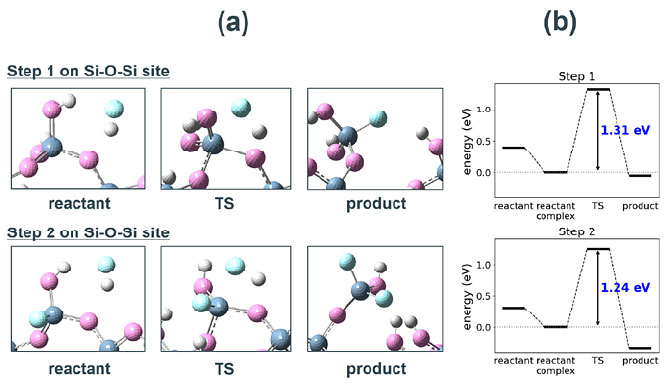 Fig. S2(a) Snapshots of the reaction process in step 1 and 2 for Si-O etching by HF on Si-O-Si site. Details are the same as in Fig. 3a.(b) Energy diagrams for step 1 and 2.
Fig. S2(a) Snapshots of the reaction process in step 1 and 2 for Si-O etching by HF on Si-O-Si site. Details are the same as in Fig. 3a.(b) Energy diagrams for step 1 and 2.モデルの妥当性の検証
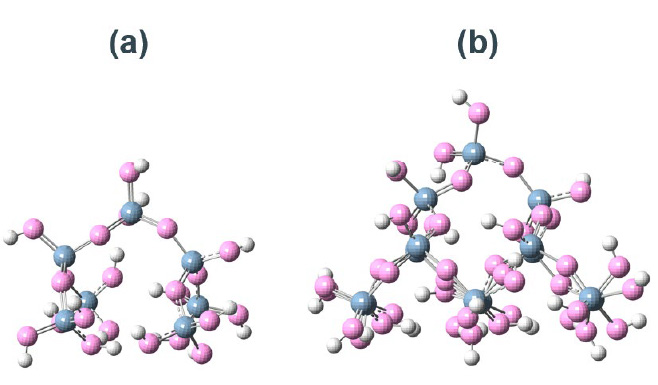 Fig. S3 Different computational models for SiO2 surfaces.(a) Si7O22H15 cluster model.(b) Si17O52H27 cluster model.
Fig. S3 Different computational models for SiO2 surfaces.(a) Si7O22H15 cluster model.(b) Si17O52H27 cluster model.

基底関数の収束性の検証
異なる基底関数で、触媒なしのHFエッチングStep1の活性化障壁は最大0.5 eV程度大きく変化することが分かった(Fig. S4)。小さい基底関数では活性化障壁を過小評価し、分極関数やdiffuse関数の追加、TZレベルの展開により改善される傾向がある。これは、HF分子の結合長の評価に起因することが分かった(Fig. S5)。あらゆる結合状態のHFにおいても、大きい基底関数と一貫性のあるHF結合距離を評価可能な最小の基底関数を調査した。汎用な1~3級アミン、エステル、アルコール、オキシド、アニオンを含む23の分子セットで、相互作用したHFの結合距離がaug-cc-pvtz基底と良い相関を示した6-31+G(d, p)基底を採用した(Fig. S6)。diffuse関数を含まない6-31G(d, p)基底では、分子種による過大・過小評価が確認された。
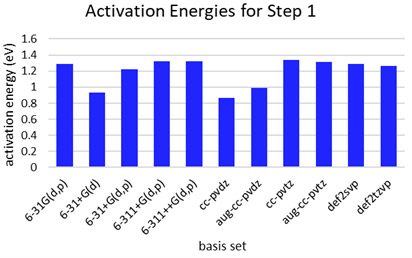 Fig. S4 Activation energies for Step 1 computed withdifferent basis set.
Fig. S4 Activation energies for Step 1 computed withdifferent basis set.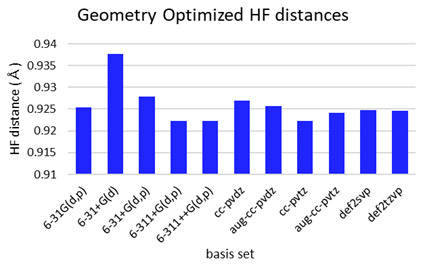 Fig. S5 Optimized HF bonding distance computed with different basis set.
Fig. S5 Optimized HF bonding distance computed with different basis set.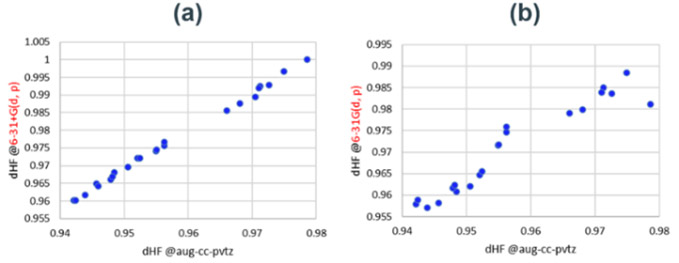 Fig. S6 HF bonding distance relative plots of structureoptimized catalyst groups with different basis set.
Fig. S6 HF bonding distance relative plots of structureoptimized catalyst groups with different basis set.(a) aug-cc-pvtz v.s. 6-31+G(d, p).
(b) aug-cc-pvtz v.s. 6-31G(d, p)
水素結合ドナーからHF分子への水素結合によるdHFの伸長
触媒-HF分子複合体の構造最適化を実行した結果、水素結合アクセプターに対してHF分子の水素原子側が吸着する場合と、同時に水素結合ドナーからフッ素原子側が吸着を受ける場合がある。これは同じ触媒基でも配座の違いによって起こりうるが、それぞれが示すdHFは大きく異なる(Fig. S7)。これは後者が、フッ素原子側の水素結合からも伸長作用を受けていることに由来する。
一方で、DFT計算で両配座の触媒活性を比較すると、大きな差がないことが分かる(Fig. S8)。これは、水素原子側の水素結合が反応過程でHF分子の伸長を続けるのに対して、フッ素原子側の水素結合はフッ素原子がケイ素原子に求核する際に切断されることが理由である。つまり、触媒活性を評価するパラメータとしてのdHFは、フッ素原子側から水素結合を受けていない構造で測定されるべきである。
データセット作成のために構造最適化した触媒-HF分子複合体の、HF分子のフッ素原子と、最も近い水素原子(以後、隣接水素原子と呼ぶ)との距離をヒストグラムに示す(Fig. S9a)。ただし、HF分子の水素原子は含まれない。フッ素原子側に水素結合を受けていない構造は、隣接水素原子との距離が3.0 Å付近にピークを持つのに対し、フッ素原子側に水素結合を受けている構造は、その強度によって1.8 Å付近までまばらに分布している。また、dHFと、隣接水素原子との距離の相関プロットからは、フッ素原子側からの強い水素結合や、複数の水素結合がdHFを大幅に増大させていることが分かる(Fig. S9b)。これらの結果から、触媒活性とdHFが相関しない構造を省くために、隣接水素原子との距離が2.4 Åより小さい構造と、HF分子のフッ素原子から3.0 Åより近くに2つ以上の水素原子が存在する構造を除外した。
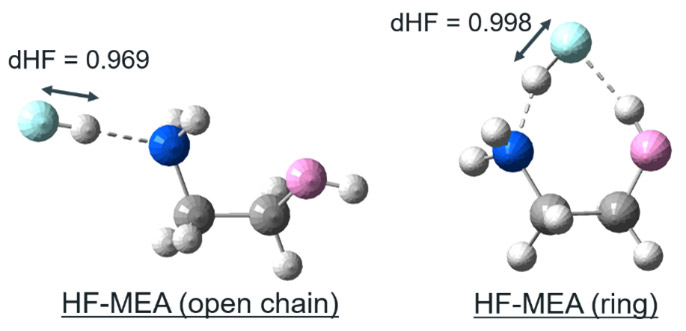 Fig. S7 Optimized structures of different conformations of MonoEthanol Amine (MEA) and dHF.
Fig. S7 Optimized structures of different conformations of MonoEthanol Amine (MEA) and dHF.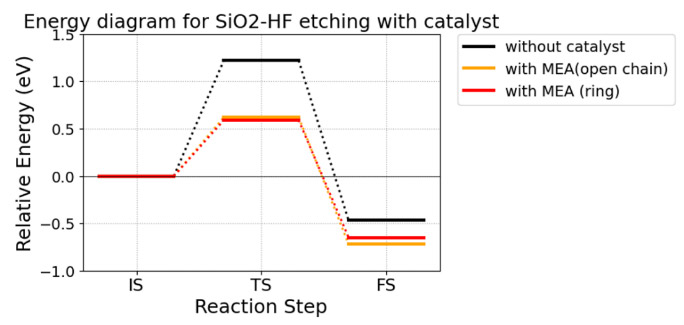 Fig. S8 Energy diagram of SiO2-HF etching catalyzed by MEA with different conformations. These ware calculated by the same method as in section 3.
Fig. S8 Energy diagram of SiO2-HF etching catalyzed by MEA with different conformations. These ware calculated by the same method as in section 3.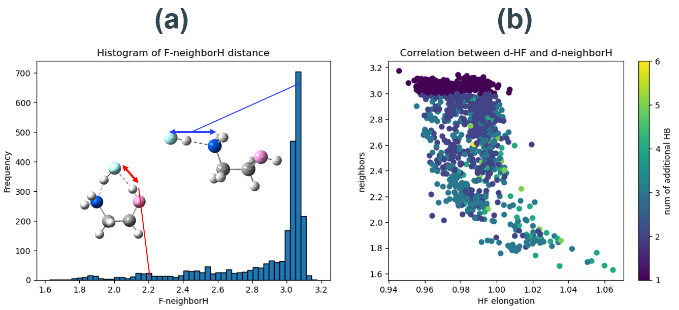 Fig. S9(a) Histograms of distances between fluorine atoms and adjacent hydrogen atoms for the structure-optimized catalyst-HF molecular complexes. As an example, the values for the two MEA in Figure S5 are explicitly shown.(b) Correlation plot between the distance between fluorine atom and adjacent hydrogen atom and dHF. The number of hydrogen atoms within 3 Å of a fluorine atom is color-coded.
Fig. S9(a) Histograms of distances between fluorine atoms and adjacent hydrogen atoms for the structure-optimized catalyst-HF molecular complexes. As an example, the values for the two MEA in Figure S5 are explicitly shown.(b) Correlation plot between the distance between fluorine atom and adjacent hydrogen atom and dHF. The number of hydrogen atoms within 3 Å of a fluorine atom is color-coded.触媒活性予測ワークフローのデータ採用条件

謝辞
本論文作成に際し、お世話になった皆様に深く感謝申し上げます。
参考文献
- Zhang, X., Zhang, Y., Zhang, X. and Guo, S.: Interface Design and Dielectric Response Behavior of SiO2/PB Composites with Low Dielectric Constant and Ultra-Low Dielectric Loss, Surf . Interfaces , 22,(2021): 100807. doi: 10.1016/j.surfin.2020.100807.
- Baek, G., Baek, J. H., Kim, H. M., Lee, S., Jin, Y., Park, H. S., Kil, D. S., Kim, S., Park, Y. and Park, J. S.: Atomic Layer Chemical Vapor Deposition of SiO2 Thin Films Using a Chlorine-free Silicon Precursor for 3D NAND Applications, Ceram . Int ., 47,(2021): 19036–19042. doi: 10.1016/j.ceramint.2021.03.249.
- Whitesides, G. M.: The Origins and the Future of Microfluidics, Nature , 442,(2006): 368–373. doi: 10.1038/ nature05058.
- Hwang, J., Cho, Y. H., Park, M. S. and Kim, B. H.: Microchannel Fabrication on Glass Materials for Microfluidic Devices, Int . J . Precis . Eng . Manuf ., 20,(2019): 479–495. doi: 10.1007/s12541-019-00103-2.
- Park, J.-S., Zhang, S., She, A., Chen, W. T., Lin, P., Yousef, K. M., Cheng, J. X. and Capasso, F.: All-glass, Large Metalens at Visible Wavelength Using Deep Ultraviolet Projection Lithography, Nano Lett ., 19,(2019): 8673–8682. doi: 10.1021/acs.nanolett.9b03333.
- Yoo, J.-H., Nguyen, H. T., Ray, N. J., Johnson, M. A., Steele, W. A., Chesser, J. M., Baxamura, S. H., Elhadj, S., McKeown, J. T., Matthews, M. J. and Feigenbaum: McKeown, J. T.; Matthews, M. J.; Feigenbaum, E. Scalable Light-printing of Substrate-Engraved Free-form Metasurfaces, ACS Appl . Mater . Interfaces , 11,(2019): 22684–22691. doi: 10.1021/acsami.9b07135.
- W i l l i a m s , K . R . a n d M u l l e r , R . S . : E t c h R a t e s f o r Micromachining Processing, Syst ., 5(4),(1996): 256–269. doi: 10.2139/ssrn.4167514.
- Bühler, J., Steiner, F. P. and Baltes, H.: Silicon Dioxide Sacrificial Layer Etching in Surface Micromachining, J . Micromech . Microeng ., 7(1),(1997): R1–R13. doi: 10.1088/0960-1317/7/1/001.
- Bliznetsov, V., Lin, H. M., Zhang, Y. J. and Johnson, D.: Deep SiO2 Etching with Al and AlN Masks for MEMS Devices, J . Micromech . Microeng ., 25(8),(2015): 087002. doi: 10.1088/0960-1317/25/8/087002.
- Chandra B. and Sudhir V.: Silicon Dioxide Films by RF Sputtering for Microelectronic and MEMS Applications, J . Micromech . Microeng ., 17(5),(2007): 1066–1077. doi: 10.1088/0960-1317/17/5/029.
- Holmes, P. J. and Snell, J. E.: A Vapor Etching Technique for the Photolithography of Silicon Dioxide, Microelectron . Reliab ., 5,(1966): 337–341. doi: 10.1016/0026-2714(66)90162-4.
- Jang, W. I., Choi, C. A., Lee, M. L., Jun, C. H. and Kim, Y. T.: Fabrication of MEMS Devices by Using Anhydrous HF Gas-Phase Etching with Alcoholic Vapor, J . Micromech . Microeng ., 12,(2002): 297–306. doi: 10.1088/0960-1317/12/3/316.
- Habuka, H. and Otsuka, T.: Reaction of Hydrogen Fluoride Gas at High Temperatures with Silicon Oxide Film and Silicon Surface, Jpn . J . Appl . Phys ., 37,(1998): 6123–6127. doi: 10.1143/JJAP.37.6123.
- Kim, C.-J., Kim, J. Y. and Sridharan, B.: Comparative E v a l u a t i o n o f D r y i n g T e c h n i q u e s f o r S u r f a c e Micromachining, Sens . Actuators A , 64(1),(1998): 17–26. doi: 10.1016/S0924-4247(98)80053-8.
- Michalik, P., Fernandez, D., Wietstruck, M., Kaynak, M. and Madrenas, J.: Experiments on MEMS Integration in 0.25 μm CMOS Process, Sensors , 18(7),(2018): 2111. doi: 10.3390/s18072111.
- Lee, Y. I., Park, K. H., Lee, J., Lee, C.-S., Yoo, H. J. and Kim, C.-J.: Dry Release for Surface Micromachining with HF Vapor-Phase Etching, J . Microelectromech . Syst ., 6(3),(1997): 226–233. doi: 10.1109/84.623111.
- Won Ick, J., Chang Auck, C., Myung Lae, L., Chi Hoon, J. and Youn Tae, K.: Fabrication of MEMS Devices by Using Anhydrous HF Gas-phase Etching with Alcoholic Vapor, J . Micromech . Microeng ., 12,(2002): 297–306. doi: 10.1088/0960-1317/12/3/316.
- Ohtake, H., Hattori, T. and Yamada, M.: Silicon Oxide Etching Mechanism by Hydrogen Fluoride and Methanol Vapor Mixture, J . Appl . Phys ., 64,(2025): 01SP15. doi: 10.35848/1347-4065/ada36e.
- Ko-hei, S., Yoshitaka, O., Ryosuke, T., Yutaka, I., Yasuo, H. and Takahiko, Y.: Atmospheric Gas-phase Catalyst Etching of SiO2 for Deep Microfabrication Using HF Gas and Patterned Photoresist, ACS Appl . Mater . Interfaces , 16,(2024): 22657–22664. doi: 10.1021/acsami.4c01291.
- Zhu, W., Zheng, G., Cao, S. and He, H.: Thermal Conductivity of Amorphous SiO2 Thin Film: A Molecular Dynamics Study, Sci . Rep ., 8,(2018): 10537. doi: 10.1038/s41598-018- 28925-6.
- Hoang, V. V.: Molecular Dynamics Simulation of Amorphous SiO2 Nanoparticles, Phys . Chem . B , 111(44),(2007): 12649–12656. doi: 10.1021/jp074237u.
- Jiao Y.-B., Wei Y.-D., Li W.-Q., Cui X.-H., Liu Z.-L., Yang J.-Q. and Li X.-J.: Threshold Displacement Energy of Amorphous SiO2: A Molecular Dynamics Study, J . Non -Cryst . Solids , 621,(2023): 122633. doi: 10.1016/j.jnoncrysol.2023.122633.
- Hoshino, T. and Nishioka, Y.: Etching Process of SiO2 by HF Molecules, J . Chem . Phys ., 111,(1999): 2109. doi: 10.1063/1.479480.
- Pettifer, R. F., Dupree, R., Farnan, I. and Sternberg, U.: NMR Determinations of Si-O-Si Bond Angle Distributions in Silica, J . Non -Cryst . Solids , 106,(1988): 408–412. doi: 10.1016/0022-3093(88)90299-2.
- Rino, J. P., Ebbsjo, I., Kalia, R. K., Nakano, A. and Vashishta, P.: Structure of Rings in Vitreous SiO2, Phys . Rev . B , 47,(1993): 3053. doi: 10.1103/PhysRevB.47.3053.
- Ramprasad, R., Batra, R., Pilania, G., Mannodi-Kanakkithodi, A. and Kim, C.: Machine Learning in Materials Informatics: Recent Applications and Prospects, npj Comput . Mater ., 3,(2017): 54. doi: 10.1038/s41524-017-0056-5.
- Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. and Walsh, A.: Machine Learning for Molecular and Materials Science, Nature , 559,(2018): 547–555. doi: 10.1038/s41586-018-0337-2.
- Toyao, T., Takahashi, K., Miyamoto, J., Mochizuki, Y., Oba, F. and Asahi, R.: Machine Learning for Catalysis Informatics: Recent Applications and Prospects, ACS Catal ., 10,(2020): 2260–2297. doi: 10.1021/acscatal.9b04186.
- Takahashi, K., Fujiwara, A., Nakanowatari, S., et al.: Catalysts Informatics: Paradigm Shift towards Datadriven Catalyst Design, Chem. Commun ., 59,(2023): 2222–2238. doi: 10.1039/D2CC05938J.
- Wu, J., Wan, Y., Wu, Z., Zhang, S., Cao, D., Hsieh, C.-Y. and Hou, T.: MF-SuP-pKa: Multi-Fidelity Modeling with Subgraph Pooling Mechanism for pKa Prediction, Acta Pharm . Sin . B , 13,(2023): 2572–2584. doi: 10.1016/j.apsb.2022.11.010.
- Johnston, R. C., Yao, K., Kaplan, Z., Chelliah, M., Leswing, K., Seekins, S., Watts, S., Calkins, D., Chief Elk, J., Jerome, S . V . , R e p a s k y , M . P . a n d S h e l l e y , J . C . : p K a a n d P r o t o n a t i o n S t a t e P r e d i c t i o n t h r o u g h M a c h i n e Learning, J . Chem. Inf . Model ., 19(8),(2023): 2380-2388. doi: 10.1021/acs.jctc.3c00044.
- Mayr, F., Wieder, M., Wieder, O. and Langer, T.: Improving Small Molecule pKa Prediction Using Transfer Learning with Graph Neural Networks, Front . Chem., 10,(2022) : 866585. doi: 10.3389/fchem.2022.866585.
- Baltruschat, M. and Czodrowski, P.: Machine Learning Meets pKa, F1000Research , 9,(2020): 113. doi: 10.12688/f1000research.22090.2.
- Scheen, J., Wu, W., Mey, A. S. J. S., Tosco, P., Mackey, M. and Michel, J.: Hybrid Alchemical Free Energy/Machine-Learning Methodology for the Computation of Hydration Free Energies, J . Chem. Inf . Model ., 60(11),(2020): 5331–5339. doi: 10.1021/acs.jcim.0c00600.
- Zhang, D., Xia, S. and Zhang, Y.: Accurate Prediction of Aqueous Free Solvation Energies Using 3D Atomic Feature-based Graph Neural Network with Transfer Learning, J . Chem. Inf . Model ., 62(8),(2022): 1840–1848. doi: 10.1021/acs.jcim.2c00260.
- Bennett, W. F. D., He, S., Bilodeau, C. L., Jones, D., Sun, D., Kim, H., Allen, J. E., Lightstone, F. C. and Ingolfsson, H. I.: Predicting Small Molecule Transfer Free Energies by Combining Molecular Dynamics Simulations and Deep Learning, J . Chem. Inf . Model ., 60(11),(2020): 5375–538. doi: 10.1021/acs.jcim.0c00318.
- Low, K., Coote, M. L. and Izgorodina, E. I.: Explainable Solvation Free Energy Prediction Combining Graph Neural Networks with Chemical Intuition, J . Chem . Inf . Model ., 62(22),(2022): 5457–5470. doi: 10.1021/acs. jcim.2c01013.
- Takahashi, A., Kumagai, Y., Miyamoto, J., Mochizuki, Y. and Oba, F.: Machine Learning Models for Predicting the Dielectric Constants of Oxides Based on High-throughput First-principles Calculations, Phys . Rev . Mater ., 4,(2020): 103801. doi: 10.1103/PhysRevMaterials.4.103801.
- Shimano, Y., Kutana, A. and Asahi, R.: Machine Learning and Atomistic Origin of High Dielectric Permittivity in Oxides, Sci . Rep ., 13,(2023): 22236. doi: 10.1038/s41598-023-49603-2.
- Saini, V., Sharma, A. and Nivatia, D.: A Machine Learning Approach for Predicting the Nucleophilicity of Organic Molecules, Phys . Chem . Chem . Phys ., 24,(2022): 1821–1829. doi: 10.1039/D1CP05072A.
- Yu, H., Wang, Y., Wang, X., Zhang, J., Ye, S., Huang, Y., Luo, Y., Sharman, E., Chen, S. and Jiang, J.: Using Machine Learning to Predict the Dissociation Energy of Organic Carbonyls, J . Phys . Chem. A, 124,(2020): 3844–3850. doi: 10.1021/acs.jpca.0c01280.
- Ho, T. K.: Random Decision Forests, Proc . 3rd Int . Conf . Doc . Anal Recognit ., 1,(1995): 278–282. doi: 10.1109/ICDAR.1995.598994.
- Breiman, L.: Random Forests, Mach . Learn ., 45,(2001): 5–32. doi: 10.1023/A:1010933404324.
- Scarselli F., Gori M., Tsoi A. C., Hagenbuchner M. and Monfardini G.: The Graph Neural Network Model, IEEE Trans . on Neu . Net ., 20(1),(2009): 61-80. doi: 10.1109/TNN.2008.2005605.
- Xie, T. and Grossman, J. C.: Crystal Graph Convolutional Neural Networks for an Accurate and Interpretable Prediction of Material Properties, Phys . Rev . Lett ., 120,(2018): 145301. doi: 10.1103/PhysRevLett.120.145301.
- S c h ü t t , K . T . , S a u c e d a , H . E . , K i n d e r m a n s , P . - J . , Tkatchenko, A. and Müller, K.-R.: SchNet - A Deep Learning Architecture for Molecules and Materials, J . Chem. Phys ., 148,(2018): 241722. doi: 10.1063/1.5019779.
- Beker, W., Roszak, R., Grybos, R. and Szymkuc, S.: Machine Learning May Sometimes Simply Capture L i t e r a t u r e P o p u l a r i t y T r e n d s : A C a s e S t u d y o f Heterocyclic Suzuki−Miyaura Coupling, J . Am . Chem . Soc ., 144,(2022): 4819–4827. doi: 10.1021/jacs.1c12005.
- Schmidt, J., Marques, M. R. G., Botti, S. and Marques, M. A. L.: Recent Advances and Applications of Machine Learning in Solid-state Materials Science, npj Comput . Mater ., 5,(2019): 83. doi: 10.1038/s41524-019-0221-0.
- Strieth-Kalthoff, F., Sandfort, F., Kühnemund, M., Kuemmel, S., Takatsu, K., Glorius, F. and Pendaries, N.: Machine Learning for Chemical Reactivity: The Importance of Failed Experiments, Angew . Chem . Int . Ed ., 61,(2022): e202204647. doi: 10.1002/anie.202204647.
- Taniike, T. and Takahashi, K.: The Value of Negative Results in Data-Driven Catalysis Research, Nat . Catal ., 6,(2023): 108–111. doi: 10.1038/s41929-023-00920-9.
- Taniike, T., Fujiwara, A., Nakanowatari, S. and Takahashi, K.: Automatic Feature Engineering for Catalyst Design Using Small Data without Prior Knowledge of Target Catalysis, Commun . Chem., 7,(2024): 11. doi: doi: 10.1038/s42004-023-01086-y.
- Becke, A. D.: Density ‐ Functional Thermochemistry. III. The Role of Exact Exchange, J . Chem. Phys ., 98,(1993): 5648–5652. doi: 10.1063/1.464913.
- Krishnan, R., Binkley, J. S., Seeger, R. and Pople, J. A.: Self‐ Consistent Molecular Orbital Methods. A Basis Set for Correlated Wave Functions, J . Chem. Phys ., 72,(1980): 650–654. doi: 10.1063/1.438955.
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A. and Nakatsuji, H., Gaussian 16 , Revision C .01 , Gaussian, Inc., Wallingford,(2016).
- The convergence criteria for geometric optimizations are as follows: the energy change per step of 1.0×10–6 hartree, the maximum force of 3.0×10–4 hartree/Bohr, the root-mean-square(RMS)values of all forces of 1.0×10–4 hartree/Bohr, the maximum atomic coordinate per step of 1.2×10–3 Bohr, and the RMS values of all atomic coordinates per step of 6.0×10–4 Bohr.
- Breneman, C. M. and Wiberg, K. B.: Determining Atomcentered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis, J . Comput . Chem ., 11(3), (1990): 361. doi: 10.1002/jcc.540110311.
- Schlegel, H. B.: Optimization of Equilibrium Geometries and Transition Structures, J . Comput . Chem., 3,(1982): 214. doi: 10.1002/jcc.540030212.
- Gonzalez, C. and Schlegel, H. B.: An Improved Algorithm for Reaction Path Following, J . Chem. Phys ., 90,(1989): 2154–2161. doi: 10.1063/1.456010.
- The IRC calculations were performed are as follows: A force constant for the calculation was determined in the initial step. A step size was 0.1(amu)1/2 Bohr and the maximum numbers of calculation times was 200.
- Bannwarth, C., Ehlert, S. and Grimme, S.: GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-binding Quantum Chemical Method with Multipole Electrostatics and Density-dependent Dispersion Contributions, J . Chem . Theory Comput ., 15(3),(2019): 1652–1671. doi: 10.1021/acs.jctc.8b01176.
- The allowable change in total energy at convergence is 5.0×10–6, the allowable change in gradient norm at convergence is 1.0×10–3, and the integral cutoff is 25.0.
- Mulliken, R. S.: Electronic Population Analysis on LCAO–MO Molecular Wave Functions., J . Chem . Phys ., 23(10),(1955): 1833–1840. doi: 10.1063/1.1740588.
- Csizmadia, I. G., Theory and Practice of MO Calculations on Organic Molecules , Elsevier,(1976).
- Schwall, K. and Zielenbach, K.: SciFinder, Chem Innov ., 30(10),(2000): 45–50. doi: 10.5195/jmla.2018.515.
- Weininger D.: SMILES, a Chemical Language and Information System. Introduction to Methodology and Encoding Rules, J . of Chem . Inf . and Com . Sci., 28,(1988): 31-36. doi: 10.1021/ci00057a005.
- RDKit, https://www.rdkit.org,(accessed April 11, 2025)
- Gasteiger, J. and Marsili, M.: A New Model for Calculating Atomic Charges in Molecules, Tetrahedron Lett ., 34,(1978): 3181–3184. doi: 10.1016/S0040-4039(01)94977-9.
- Gasteiger J, Marsili M.: Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges, Tetrahedron ., 36(22)(1980):3219–3228. doi: 10.1016/0040-4020(80)80168-2.
- Kursa, M., Jankowski, A. and Rudnicki, W.: Boruta – A System for Feature Selection, Fund . Inform., 101,(2010): 271–285. doi: 10.3233/FI-2010-2.
- Pedregosa, F.: Scikit-learn: Machine Learning in Python, J . Mach . Learn . Res ., 12,(2011): 2825–2830. doi: 10.1016/j.patcog.2011.04.006.
- Hoerl, A. and Kennard, R.: Ridge Regression: Biased Estimation for Nonorthogonal Problems, Technometrics , 12,(1970): 55–67. doi: 10.2307/1267351.
- Tibshirani, R.: Regression Shrinkage and Selection Via the Lasso, J . Roy . Stat . Soc . B , 58,(1996): 267–288. doi: 10.1111/j.2517-6161.1996.tb02080.
- Moriwaki, H., Tian, Y.-S., Kawashita, N. and Takagi, T.: M o r d r e d : A M o l e c u l a r D e s c r i p t o r C a l c u l a t o r , J . Cheminformatics , 10,(2018): 4. doi; 10.1186/s13321-018-0258-y.
- Grimme, S., Bannwarth, C. and Shushkov, P.: A Robust and Accurate Tight-binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All spd-Block Elements(Z=1–86), J . Chem . Theory Comput ., 13(5),(2017): 1989–2009. doi: 10.1021/acs. jctc.7b00118.
- Bannwarth, C., Caldeweyher, E., Ehlert, S., Hansen, A., Pracht, P., Seibert, J., Spicher, S. and Grimme, S.: E x t e n d e d T i g h t - b i n d i n g Q u a n t u m C h e m i s t r y Methods, WIREs Comput . Mol . Sci ., 11,(2021): e1493. doi: 10.1002/wcms.1493.
- Dewar, M. J. S., Zoebisch, E. G., Healy, E. F. and Stewart, J. J. P.: Development and Use of Quantum Mechanical Molecular Models: A New General Purpose Quantum Mechanical Molecular Model, J . Am . Chem . Soc ., 107,(1985): 3902–3909. doi: 10.1021/ja00299a024.
- Stewart, J. J. P.: Optimization of Parameters for Semiempirical Methods: More Modifications to the NDDO Approximations and Re-Optimization of Parameters, J . Mol . Model ., 19,(2013): 1–32. doi: 10.1007/s00894-012-1667.
- Grimme, S., Muller, M. and Hansen, A.: A Non-selfconsistent Tight-binding Electronic Structure Potential in a Polarized Double-ζ Basis Set for All Spd-Block Elements up to Z = 86, J . Chem. Phys ., 158,(2023): 124111. doi: 10.1063/5.0137838.
- Verma, S., Rivera, M., Scanlon, D. O. and Walsh, A.: Machine Learned Calibrations to High-Throughput Molecular Excited State Calculations, J . Chem . Phys ., 156,(2022): 134116. doi: 10.1063/5.0084535.
- Abarbanel, O. D. and Hutchison, G. R.: QupKake: Integrating Machine Learning and Quantum Chemistry for Micro-pKa Predictions, J . Chem . Theory Comput ., 20(15),(2024): 6946–6956. doi: 10.1021/acs.jctc.4c00328.
- Matsubara, K., Takahashi, K., Matsuda, T., Ueki, Y., Seko, N. and Kakuchi, R.: GFN-xTB-based Computations Provide Comprehensive Insights into Emulsion Radiation-Induced Graft Polymerization, Chem . Plus Chem ., 89(4),(2024): e202300480. doi: 10.1002/cplu.202300480.
- Hengl, T., Nussbaum, M., Wright, M. N., Heuvelink, G. B. M . a n d G r ä l e r , B . : R a n d o m F o r e s t a s A G e n e r i c Framework for Predictive Modeling of Spatial and Spatio-Temporal Variables, PeerJ , 6,(2018): e5518. doi: 10.7717/peerj.5518.
- Holland, J. H., Adaptation in Natural and Artificial Systems , University of Michigan Press, Ann Arbor, MI, 1975.
- Fogel, D. B., Evolutionary Computation : The Fossil Record ,(1998)
- G o l d b e r g , D . E . , G e n e t i c A l g o r i t h m s i n S e a r c h , Optimization , and Machine Learning ,(1989)
- Carstensen, N. O., Dieterich, J. M. and Hartke, B.: Design of O p t i m a l l y S w i t c h a b l e M o l e c u l e s b y G e n e t i c Algorithms, Phys . Chem. Chem. Phys ., 13,(2011): 2903–2910. doi: 10.1039/c0cp01065k.
- Carlotto, S., Orian, L. and Polimeno, A.: Heuristic Approaches to the Optimization of Acceptor Systems in Bulk Heterojunction Cells: A Computational Study, Theor . Chem. Acc ., 131,(2012): 1191. doi: 10.1007/s00214-012-1191-1.
- Chen, Z., Li, D., Liu, M. and Liu, J.: Graph Neural Networks with Molecular Segmentation for Property Prediction and Structure–property Relationship Discovery, Comput . Chem. Eng ., 179,(2023): 108403. doi: 10.1016/j.compchemeng.2023.108403.
- Hiroaki I., Ryosuke T., Tomoaki S. and Yoshitaka O.: Development of Highly Catalytic Polymers for Highspeed HF-Dry-etching of Quartz-based Materials, AGC研究開発報告, 6174,(2024)
- Ying-Hung S., Jerry P. H. and Cheryl L. M.: A Mechanistic Study of Polybenzoxazole Formation with Model Compounds, Macromolecules , 28(21),(1995): 7289–7290. doi: 10.1021/ma00125a038.
- Wolfe J. F. and Arnold F.: Rigid-rod Polymers. Synthesis and Thermal Properties of Para-aromatic Polymers with 2,6-Benzobisthiazole Units in the Main Chain, Macromolecules , 14(4)(1981): 909-915. doi: 10.1021/ma50005a005.
- Choe E. W. and Kim S. N.: Synthesis, Spinning, and Fiber Mechanical Properties of Poly(p-Phenylenebenzobisoxazole), Macromolecules , 14(4)(1981): 920-924. doi: 10.1021/ma50005a006.
- Kenichi F. and Minoru U.:感光性ポリイミド・感光性ポリベンズオキサゾールの開発, Kobunshi Ronbunshu , 63(9),(2006): 561-576. doi: 10.1295/koron.63.561.doi
- Hean Z., Shaohua J., Gaigai D., Juanhua L., Kunming L., Caiyun Z., Haoqing H.: Heat-resistant Polybenzoxazole Nanofibers Made by Electrospinning, Eur . Poly . J ., 50,(2014): 61-68. doi: 10.1016/j.eurpolymj.2013.10.029.
- McInnes, L., Healy, J., Saul, N. and Groberger, L.: UMAP: Uniform Manifold Approximation and Projection, J . Open Source Softw ., 3(29),(2018): 861. doi: 10.21105/joss.00861.